




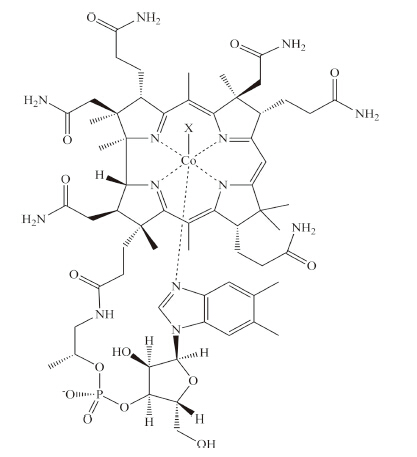
Vitamin B12 (cyanocobalamin,CNCbl) and its derivatives are essential human nutrients,originally discovered as a treatment for pernicious anemia [1, 2, 3, 4, 5]. The B12 vitamers (a vitamer is one of a class of related chemical substances that fulfill the same specific vitamin function) are cobalt centered corrinoid rings featuring four equatorial nitrogen ligands provided by the ring along with variable upper and lower axial ligands. The electronic structure and chemical function of cobalamin compounds are controlled by the axial ligands. In solution and in some enzymes,for example ethanolamine deaminase [6],the lower axial ligand is a dimethylbenzimidazole group attached to the corrin ring through an alkylphosphate tail (Fig. 1). In a number of other enzymes, including the human enzymes methylmalonyl Co-A mutase [7] and methionine synthase [8],the dimethylbenzimidazole ligand is replaced by a histidine residue. The lower axial ligand can also be replaced by a water molecule resulting in a base-off cobalamin compound. This base-off form dominates in solution at low pH [9], in certain transport proteins [10] and a small number of methyl transferase proteins [2]. The upper axial ligand is an alkyl group in the enzymatically active B12 coenzymes. Methylcobalamin (MeCbl) catalyzes methyl transfer reactions in methionine synthase while 50-deoxyadenosylcobalamin (AdoCbl) catalyzes radical rearrangement reactions in a variety of enzymes including human methylmalonyl Co-A mutase [5].

|
Download:
|
| Fig. 1. Cobalamin cofactor. The upper axial ligand,designated X,can be an alkyl or non-alkyl group as discussed in the text. | |
{kind=link}
In addition to the native biological function,cobalamin cofactors have recently received increased attention as a vehicle for optically controlled drug delivery and as a source of hydroxyl radicals [11, 12] and as antivitamins [13, 14]. The effective development of photoactive cobalamin compounds requires a detailed understanding of the intramolecular and intermolecular factors that control their photochemistry and photophysics. It is helpful to begin with the simplest cofactors and progress through to the more complex cobalamin compounds. Here we review the current state of understanding arising from ultrafast spectroscopic studies and theoretical simulations for the simplest non-alkylcobalamin CNCbl and present new broadband transient absorption measurements on CNCbl as a function of solvent. 2. Background
The electronic structure and chemical function of cobalamin compounds are controlled by the axial ligands. Ultrafast broadband transient absorption spectroscopy has been used to study the photochemistry and photophysics of naturally occurring and synthetically prepared cobalamin compounds [9, 15, 16, 17, 18, 19, 20, 21, 22, 23, 24, 25, 26, 27]. Alkylcobalamins are photolabile. For most alkylcobalamins that have been studied to date,the primary photolysis quantum yield is on the order of unity [15, 21, 22, 24]. The quantum yield for long-lived radicals is determined by competition between diffusive separation of the radical pair and geminate recombination [15]. Methylcobalamin is an exception with a pronounced wavelength dependence to the primary photolysis quantum yield [19, 23, 25]. Derivatives containing non-alkyl upper axial ligands (such as cyano-,hydroxo-,aquo-,and azido-) present markedly different photochemistry [16, 17, 26]. The photoexcitation of non-alkyl cobalamins is characterized by formation of short-lived electronically excited states that recover to the ground state on a picosecond timescale with little to no photolysis quantum yield.
Time dependent density functional theory (TD-DFT) studies by Kozlowski and coworkers have identified a triplet sigma antibonding state that was suggested to mediate the photolysis of alkyl cobalamins [28, 30, 31, 32, 33, 34]. Detailed comparison of MeCbl and ethylcobalamin (EtCbl) led to the suggestion that changes in the barrier for crossing from the S1 state to the dissociative channel accounted for the distinctive wavelength dependence and photolysis quantum yield following excitation of MeCbl. A more recent calculation exploring the S1 potential energy surface has called this into question,suggesting instead that a ligand field state plays the crucial role in controlling the photodissociation [29]. In the non-alkyl cobalamins,the dissociation channel is much higher in energy and shifted far from the Franck-Condon region and thus is a non-relevant excited state pathway [30, 35, 36, 37, 38]. 3. The S1 state of CNCbl
Vitamin B12,CNCbl,provides a paradigm system for the investigation of nonalkyl cobalamins. Careful transient absorption measurements combined with detailed theoretical simulations have illuminated many of the details of the electronic structure controlling the photochemistry and photophysics.
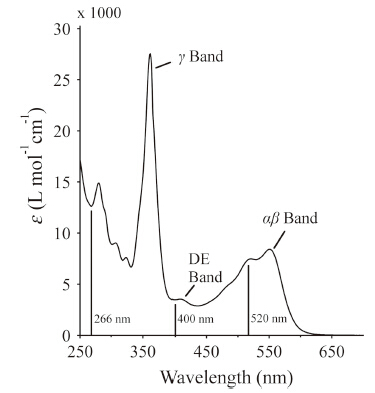
Ultrafast spectroscopic methods including broadband visible transient absorption spectroscopy and infrared spectroscopy have been used to study the photochemistry and photophysics of CNCbl after excitation of the DE and αβ bands (Fig. 2) [16, 17]. The bandwidth of the probe in the broadband visible transient absorption experiments ranged from ca. 450-700 nm,allowing global kinetic analysis of the decay of the excited state absorption and recovery of the αβ band ground state bleach. The dynamics of the excited state absorption are characterized by one or two subpicosecond components followed by a solvent-dependent ca. 10 ps decay of the transient absorption signal [16, 17, 26]. The subpicosecond components account for rapid internal conversion from the initially excited electronic state to the lowest energy excited state,S1. The solvent-dependent picosecond lifetime of the S1 state reflects the influence of a small barrier along the pathway for internal conversion from the S1 state to the ground state.

|
Download:
|
| Fig. 2. Absorption spectrum of CNCbl in water. The vertical lines at 266 nm,400 nm, and 520 nm indicate excitation wavelengths used in ultrafast spectroscopic measurements. | |
{kind=link}
The ultrafast electronic relaxation of CNCbl has also been measured for the gas phase isolated molecule using excitation at 400 nm followed by 800 nm multiphoton ionization or excitation at 266 nm followed by 400 nm multiphoton ionization [27]. The signals obtained in both measurements are characterized by a strong ionization signal at early times. Following excitation at 400 nm this signal decays to baseline on a 100 ± 30 fs time scale. Following excitation at 266 nm the ionization signal decays on a 100 ± 10 fs time scale with an additional small amplitude signal (5%) decaying on a longer picosecond time scale. The data reflects fast internal conversion from the initially excited state to a relaxed state, presumably the S1 state. This fast internal conversion is consistent with the time scales observed in solution. The low intensity of the picosecond evolution is attributed to the nature of the experiment. Ionization of the S1 state will require an additional photon.
Time-resolved IR (TRIR) measurements probing the C≡N triple bond in ethanol are characterized by a significant red-shift of the C≡N stretching vibration from 2138 cm-1 in the ground state to 2120 cm-1 in the S1 state [17]. This frequency is significantly higher than CN- at 2080 cm-1 consistent with a C-Co bond that is weakened,but not broken,in the S1 excited state. The visible absorption of the S1 state between 460 nm and 640 nm is characterized by a blue shift of the αβ band similar to that observed for base-off alkylcobalamins [17]. A blue shift of the αβ band is also observed for cyanocobinamide [39]. Thus the infrared absorption suggests a weakening of the C-Co bond and the shift in the electronic absorption spectrum is consistent with a weakening of the axial Co-N bond. As a result the S1 state was tentatively assigned as a π→ σ*(dz2) ligand-to-metal charge transfer (LMCT) state. More recently,a TRIR measurement of CNCbl in D2O was performed in the fingerprint region from 1700 cm-1 to 1300 cm-1 [26]. The TRIR spectrum is characterized by dominant ground state bleach signals at 1640 cm-1 (shifting on a picosecond time scale to 1626 cm-1),1575 cm-1,1504 cm-1,and ca. 1360 cm-1,accompanied by small absorption increases at other frequencies. The most significant absorption increase is at ca. 1425 cm-1. The precise interpretation of these changes in the vibrational spectrum is confounded by the complicated mode assignments for each vibrational band in the congested IR spectrum. However,most of the vibrations active in the TRIR spectrum involve some degree of motion of the corrin ring,which is consistent with an S1 state involving excitation of an electron from the p HOMO orbital of CNCbl.
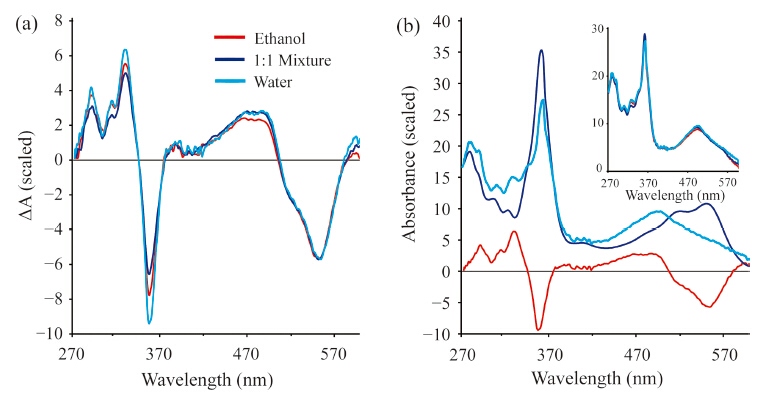
The transient absorption experiments reported earlier did not extend into the UV and were unable to probe the dynamics of the γ band at ca. 350 nm. The γ band is qualitatively indicative of the upper axial ligand field strength. Thus transient spectra in this spectral range may shed additional light on the nature of the excited electronic state. It is now possible to obtain high-quality transient spectra that span the region from 270 nm to 600 nm. We have obtained such transient spectra for CNCbl in water,ethanol, and a 1:1 mix of water and ethanol. The excitation wavelength for these measurements was 266 nm to avoid interference from scatter of the pump pulse. These transient spectra are characterized by rapid internal conversion to the S1 state on a time scale <200 fs followed by decay to the ground state on time scales consistent with those reported following excitation at 400 nm and 520 nm. The S1 state spectrum decays on a 6.5 ps time scale in water,a 10 ps time scale in the 1:1 mixed solvent,and a 14 ps time scale in ethanol. The difference spectra of the S1 states in each solvent system are compared in Fig. 3(a).

|
Download:
|
| Fig. 3. (a) Difference spectra obtained following excitation of CNCbl at 266 nm in water,ethanol,and a 1:1 mixture of water and ethanol. The spectra are integrated over the S1 lifetime and normalized to the same value in the region around 550 nm for comparison. (b) Estimation of the S1 spectrum in water. The ground state spectrum multiplied by a parameter,α,(dark blue) is added to the experimental difference spectrum (red) to obtain the estimated excited state spectrum (light blue). The estimated spectra in all three solvents are compared in the inset: water (light blue),1:1 mixture (dark blue),and ethanol (red). | |
{kind=link}
It is possible to estimate the absorption spectrum of the S1 state, ES(λ),by adding the ground state spectrum,GS(λ),back into the difference spectrum,DS(λ) using a parameter a: ES(λ) = DS(λ) + a GS(λ) [17]. The estimated spectrum for the S1 state in water is plotted in Fig. 3(b) with the spectra in all three solvents compared in the inset. The blue shift of the αβ band is consistent with the spectra reported following 400 nm excitation,but the current data set provides a more complete picture of the excited state spectrum. The S1 γ band is weaker than the ground state γ band and is slightly red-shifted from the ground state band by ~1 to 2 nm. The lack of solvent dependence on the S1 spectrum implies that the nature of the S1 state is unaffected by the solvent polarity. Thus,the solvent polarity does not modify the structure of the electronically excited CNCbl molecule. The small red-shift in the γ band runs counter to the 7 nm blue-shift observed for ground state cyanocobinamide with water as the lower axial ligand [39]. This may reflect the combined influence of both axial ligands and a change in the electronic configurations responsible for the transition in the S1 state [30, 38].
The rate of decay of the S1 state correlates with the dielectric constant of the solvent,but not with solvent viscosity [16, 17]. This suggests that the S1 state has significant LMCT character,allowing for stabilization by polar solvents. Temperature dependent measurements in water and ethylene glycol were modeled using a modified Arrhenius expression [16]:
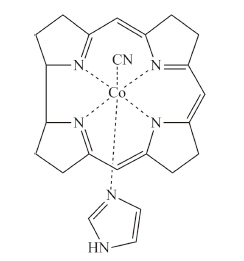
Detailed quantum chemical calculations have been performed for a simplified CNCbl model (Fig. 4) using TD-DFT methods with an implicit COSMO solvent model,the BP86 functional and the TZVPP basis set [30, 35, 37, 38]. Excitation of CNCbl results in the initial Franck-Condon population of any of a number of states dominated by π→π* excitation of the corrin ring. The specific state is determined by the excitation wavelength. Fast internal conversion from the initially excited state results in formation of a π→d LMCT state with a geometry similar to the ground state. Subsequent internal conversion to the S1 π→σ*(dz2) LMCT state involves elongation of the C-Co and Co-Naxial bonds. In the minimum energy structure the C-Co bond elongates by ca. 19% from 1.9Å to 2.2Å and the Co-Naxial bond elongates by ca. 10% from 2.05Å to 2.27Å . Internal conversion back to the ground state occurs via a pathway involving further elongation of the C-Co bond to ca. 2.65Å to reach the S1/S0 crossing. The barrier for the internal conversion process is ~5 kcal/mol. This barrier is in reasonable agreement with the 2.1 kcal/mol barrier in water deduced from the temperature dependent transient absorption measurements. The overall agreement between the experimental measurements and the TD-DFT calculations is quite good leading to a clear picture of the electronic structure and excited state dynamics.

|
Download:
|
| Fig. 4. Model CNCbl compound used in the TD-DFT calculations. | |
{kind=link}
Vitamin B12,CNCbl,is a paradigm molecule for elucidation of the electronic structure of complex biological cofactors. The comparison of high level theoretical calculations with high quality transient absorption measurements allows for detailed analysis of the structure of the electronically excited state. The spectral changes are characteristic of structural changes involving elongation of the axial bonds. This agrees with structural changes predicted by the TD-DFT calculations. The experimental and calculated barriers for internal conversion and ground state recovery are in acceptable agreement given the potential errors in the experiment and the limitations of the computational methods. These ultrafast transient absorption experiments and theoretical simulations of CNCbl lay the groundwork for detailed studies of a range of cobalamin cofactors proposed for use as anti-vitamins,photoactivated drug delivery agents,and in situ production of hydroxyl radicals. 5. Materials and methods
A KM Labs Ti:Sapphire oscillator was used to generate an 88 MHz pulse train of ca. 800 nm pulses with a bandwidth of ca. 30 nm. The 88 MHz pulse train was reduced to 1 kHz pulse train by a pulse picker (Quantum Technologies) and subsequently amplified by a tandem regenerative amplifier and 3 pass amplifier, resulting in a 1W average power beam. This beam was divided between the pump (~100 mW) and probe (~50 mW) arms of the transient absorption setup using a variable attenuator. A 266 nm pump beam was generated by frequency tripling 800 nm using two β barium borate (BBO) crystals. The pulses were maintained at ca. 250 nJ. A λ/2 plate was used to rotate the polarization of the pulses to 54.78 (magic angle) with respect to the probe beam to remove dipole reorientation contributions from the signal resulting. A chopper operating at 125 Hz (4 on and 4 off) modulated the pump pulses to produce the pump on and pump off probe spectra. The probe consisted of a continuum (280-600 nm) generated by focusing the second harmonic of the laser (400 nm) into a 5 mm calcium fluoride (CaF2) plate. Pump/probe time delays were achieved via mechanical delay line. The CaF2 was continuously translated during the experiments to prevent optical damage. A solution of nickel sulfate in a 1 mm cuvette was used to filter out the residual 400 nm. The continuum was focused and overlapped with the pump in the sample using a spherical mirror. The probe was detected by a diode array spectrometer (Avantes). All reagents used in these experiments were obtained from Sigma-Aldrich and used without purification. Transient absorption measurements were performed on solutions of CNCbl in water,ethanol,and a 1:1 mixture of water and ethanol. The CNCbl concentration was ~0.7 mmol/L to ensure an optical density of 1.0 at 266 nm with 1 mmpath length quartz flow cells (NSG) used for all experiments.
AcknowledgmentsThis work was supported in part by NSF CHE-1150660. We also thank Prof. Kenneth Spears for helpful conversations and assistance with the laser system.
| [1] | R. Banerjee, S.W. Ragsdale, The many faces of vitamin B12: catalysis by cobalamin- dependent enzymes, Ann. Rev. Biochem. 72 (2003) 209-247. |
| [2] | R.G. Matthews, M. Koutmos, S. Datta, Cobalamin-dependent and cobamidedependent methyltransferases, Curr. Opin. Struct. Biol. 18 (2008) 658-666. |
| [3] | M.L. Ludwig, R.G. Matthews, Structure-based perspectives on B12-dependent enzymes, Ann. Rev. Biochem. 66 (1997) 269-313. |
| [4] | E.N.G. Marsh, Coenzyme B12 (cobalamin)-dependent enzymes, Essays Biochem. 34 (1999) 139-154. |
| [5] | R. Banerjee, Chemistry and Biochemistry of B12, John Wiley and Sons, New York, 1999. |
| [6] | S.C. Ke, K. Warncke, Interactions of substrate and product radicals with CoII in cobalamin and with the active site in ethanolamine deaminase, characterized by ESE-EPR and 14N ESEEM spectroscopies, J. Am. Chem. Soc. 121 (1999) 9922-9927. |
| [7] | F. Mancia, N.H. Keep, A. Nakagawa, et al., How coenzyme B12 radicals are generated: the crystal structure of methylmalonyl-coenzyme A mutase at 2 a? resolution, Structure 4 (1996) 339-350. |
| [8] | C.L. Drennan, S. Huang, J.T. Drummond, R.G. Matthews, M.L. Ludwig, How a protein binds B12: a 3.0 A X-ray structure of B12-binding domains of methionine synthase, Science 266 (1994) 1669-1674. |
| [9] | J.A. Peng, K.C. Tang, K. McLoughlin, et al., Ultrafast excited-state dynamics and photolysis in base-off B-12 coenzymes and analogues: absence of the transnitrogenous ligand opens a channel for rapid nonradiative decay, J. Phys. Chem. B 114 (2010) 12398-12405. |
| [10] | D. Padovani, T. Labunska, B.A. Palfey, D.P. Ballou, R. Banerjee, Adenosyltransferase tailors and delivers coenzyme B12, Nat. Chem. Biol. 4 (2008) 194-196. |
| [11] | T.A. Shell, J.R. Shell, Z.L. Rodgers, D.S. Lawrence, Tunable visible and near-IR photoactivation of light-responsive compounds by using fluorophores as lightcapturing antennas, Angew. Chem. Int. Ed. 53 (2014) 875-878. |
| [12] | T.A. Shell, D.S. Lawrence, A new trick (hydroxyl radical generation) for an old vitamin (B12), J. Am. Chem. Soc. 133 (2011) 2148-2150. |
| [13] | M. Ruetz, R. Salchner, K. Wurst, S. Fedosov, B. Kra¨ utler, Phenylethynylcobalamin: a light-stable and thermolysis-resistant organometallic vitamin B12 derivative prepared by radical synthesis, Angew. Chem. Int. Ed. 52 (2013) 11406-11409. |
| [14] | E. Mutti, M. Ruetz, H. Birn, B. Krautler, E. Nexo, 4-ethylphenyl-cobalamin impairs tissue uptake of vitamin B-12 and causes vitamin B12 deficiency in mice, PLOS ONE 8 (2013) e75312. |
| [15] | A. Stickrath, E.C. Carroll, X. Dai, et al., Solvent-dependent cage dynamics of small nonpolar radicals: lessons from the photodissociation and geminate recombination of alkylcobalamins, J. Phys. Chem. A 113 (2009) 8513-8522. |
| [16] | D.A. Harris, A.B. Stickrath, E.C. Carroll, R.J. Sension, Influence of environment on the electronic structure of Cob(III) alamins: time-resolved absorption studies of the s1 state spectrum and dynamics, J. Am. Chem. Soc. 129 (2007) 7578-7585. |
| [17] | J.J. Shiang, A.G. Cole, R.J. Sension, et al., Ultrafast excited-state dynamics in vitamin B12 and related Cob(III) alamins, J. Am. Chem. Soc. 128 (2006) 801-808. |
| [18] | R.J. Sension, D.A. Harris, A. Stickrath, et al., Time-resolved measurements of the photolysis and recombination of adenosylcobalamin bound to glutamate mutase, J. Phys. Chem. B 109 (2005) 18146-18152. |
| [19] | R.J. Sension, D.A. Harris, A.G. Cole, Time-resolved spectroscopic studies of B12 coenzymes: a comparison of the influence of solvent on the primary photolysis mechanism and geminate recombination of methyl-, ethyl-, n-propyl-, and 50- deoxyadenosylcobalamin, J. Phys. Chem. B 109 (2005) 21954-21962. |
| [20] | R.J. Sension, A.G. Cole, A.D. Harris, et al., Photolysis and recombination of adenosylcobalamin bound to glutamate mutase, J. Am. Chem. Soc. 126 (2004) 1598-1599. |
| [21] | A.G. Cole, L.M. Yoder, J.J. Shiang, et al., Time-resolved spectroscopic studies of B12 coenzymes: a comparison of the primary photolysis mechanism in methyl-, ethyl-, n-propyl-, and 50-deoxyadenosylcobalamin, J. Am. Chem. Soc. 124 (2002) 434-441. |
| [22] | L.M. Yoder, A.G. Cole, L.A. Walker II, R.J. Sension, Time-resolved spectroscopic studies of B12 coenzymes: influence of solvent on the photolysis of adenosylcobalamin, J. Phys. Chem. B 105 (2001) 12180-12188. |
| [23] | J.J. Shiang, L.A. Walker II., N.A. Anderson, A.G. Cole, R.J. Sension, Time-resolved spectroscopic studies of B12 coenzymes: the photolysis of methylcobalamin is wavelength dependent, J. Phys. Chem. B 103 (1999) 10532-10539. |
| [24] | L.A. Walker II., J.J. Shiang, N.A. Anderson, S.H. Pullen, R.J. Sension, Time-resolved spectroscopic studies of B12 coenzymes: the photolysis and geminate recombination of adenosylcobalamin, J. Am. Chem. Soc. 120 (1998) 7286-7292. |
| [25] | L.A. Walker II., J.T. Jarrett, N.A. Anderson, et al., Time-resolved spectroscopic studies of B12 coenzymes: the identification of a metastable Cob(III)alamin photoproduct in the photolysis of methylcobalamin, J. Am. Chem. Soc. 120 (1998) 3597-3603. |
| [26] | A.R. Jones, H.J. Russell, G.M. Greetham, et al., Ultrafast infrared spectral fingerprints of vitamin B12 and related cobalamins, J. Phys. Chem. A 116 (2012) 5586-5594. |
| [27] | N. Shafizadeh, L. Poisson, B. Soep, Ultrafast electronic relaxation of excited state vitamin B12 in the gas phase, Chem. Phys. 350 (2008) 2-6. |
| [28] | K. Kornobis, N. Kumar, P. Lodowski, et al., Electronic structure of the S1 state in methylcobalamin: Insight from CASSCF/MC-XQDPT2, EOM-CCSD, and TD-DFT calculations, J. Comp. Chem. 34 (2013) 987-1004. |
| [29] | P. Lodowski, M. Jaworska, T. Andruniow, B.D. Garabato, P.M. Kozlowski, Mechanism of Co-C bond photolysis in the base-on form of methylcobalamin, J. Phys. Chem. A 118 (2014) 11718-11734. |
| [30] | H. Solheim, K. Kornobis, K. Ruud, P.M. Kozlowski, Electronically excited states of vitamin B12 and methylcobalamin: theoretical analysis of absorption, CD, and MCD data, J. Phys. Chem. B 115 (2011) 737-748. |
| [31] | P. Lodowski, M. Jaworska, T. Andruniów, M. Kumar, P.M. Kozlowski, Photodissociation of Co-C bond in methyl- and ethylcobalamin: an insight from TD-DFT calculations, J. Phys. Chem. B 113 (2009) 6898-6909. |
| [32] | T. Andruniów, M. Jaworska, P. Lodowski, et al., Time-dependent density functional theory study of cobalt corrinoids: electronically excited states of coenzyme B12, J. Chem. Phys. 131 (2009) 105105. |
| [33] | T. Andruniów, M. Jaworska, P. Lodowski, et al., Time-dependent density functional theory study of cobalt corrinoids: electronically excited states of methylcobalamin, J. Chem. Phys. 129 (2008) 085101. |
| [34] | M. Jaworska, P. Lodowski, T. Andruniów, P.M. Kozlowski, Photolysis of methylcobalamin: identification of the relevant excited states involved in Co-C bond scission, J. Phys. Chem. B 111 (2007) 2419-2422. |
| [35] | P. Lodowski, M. Jaworska, T. Andruniów, B.D. Garabato, P.M. Kozlowski, Mechanism of the S1 excited state internal conversion in vitamin B12, Phys. Chem. Chem. Phys. 16 (2014) 18675-18679. |
| [36] | M. Kumar, P.M. Kozlowski, Why hydroxocobalamin is photocatalytically active? Chem. Phys. Lett. 543 (2012) 133-136. |
| [37] | P. Lodowski, M. Jaworska, K. Kornobis, T. Andruniów, P.M. Kozlowski, Electronic and structural properties of low-lying excited states of vitamin B12, J. Phys. Chem. B 115 (2011) 13304-13319. |
| [38] | K. Kornobis, N. Kumar, B.M. Wong, et al., Electronically excited states of vitamin B12: benchmark calculations including time-dependent density functional theory and correlated ab initio methods, J. Phys. Chem. A 115 (2011) 1280-1292. |
| [39] | M.S.A. Hamza, J.M. Pratt, The chemistry of vitamin B12. Part 29. Coordination of imidazoles and 1 2,4-triazole by aquacyanocobinamide, J. Chem. Soc. Dalton Trans. (1994) 1373-1376. |