




b LSA Biophysics, University of Michigan, Ann Arbor, MI 48109, USA
Hydrophobicity is an important driver of the biomacromolecular structures that ultimately determine much of the function of cellular machinery. The current thermodynamic picture of hydrophobic hydration is size-dependent: small hydrophobes allow water to maintain its hydrogen bonding network by adopting a somewhat restricted set of conformations,resulting in a largely entropic solvation free energy cost [1]. Large hydrophobes,on the other hand,cause water to sacrifice hydrogen bonds,yielding an enthalpic solvation free energy. The transition from small to large hydrophobe size occurs at a length scale of roughly 1 nm,which is the characteristic dimension of proteins. Hence,a detailed understanding of the energetics and dynamics of hydration should be helpful in describing quantitative phenomena in biology.
Using ultrafast 2D-IR spectroscopy coupledwith simulations,we approachedbiomoleculehydration firstby characterizing the nature of water at the protein surface in an isolated environment [2, 3].We then modified the degree of hydration using trifluoroethanol to displace water heterogeneously,and altered the solvent’s dynamics alone using glycerol [3, 4, 5]. To approach a cellular environment,we studied the effects of macromolecular crowders on the hydration and protein dynamics [6].
Much of the theoretical and experimental work on biomolecule hydration aims to describe free energies and solvation structure. It is essential to be able to rationalize or predict where water molecules are and the energetic cost of removing them,since many processes,including protein folding,ligand binding,protein- protein interactions,and membrane formation,depend crucially on water [7]. In considering the dynamics of biomolecular hydration,however,it might be less clear what is to be gained. Although it is widely acknowledged that water should behave somewhat differently at interfaces,the dynamical motion of water is still much faster than the relevant biological time scales associated with,for example,enzyme catalysis or diffusive motion. Nevertheless,diffusion on any length or time scales results from numerous microscopic collisions that necessarily take place on ultrafast time scales. Indeed,substrate binding to active sites,as well as protein folding and protein-protein complexation are processes that fundamentally include water motion.
It is challenging to measure the dynamics of water near a biomolecule surface. We focus here on our new method combining small transition metal complexes used as probes in conjunction with ultrafast two-dimensional infrared spectroscopy. These probes can be studied either directly in solution,covalently attached to proteins,or inserted into membrane bilayers by conjugation to lipids or cholesterol [2, 3, 4, 6, 8]. We review here our studies of small carbon monoxide releasing molecules (CORMs), metal-carbonyl labeled proteins and membranes. 2. Two-dimensional infrared (2D-IR) spectroscopy
In analogy with multidimensional NMR spectroscopy,which correlates an excited frequency with a detected frequency,2D-IR spectroscopy (Fig. 1A) provides a frequency map that enables direct measurements of vibrational coupling,energy transfer and relaxation,as well as spectral dynamics and line shape details. 2DIR spectroscopy in general contains rich chemical information,but for the purposes of this short review,we focus on only one aspect that is particularly relevant to hydration dynamics. 2D spectroscopy exposes spectral inhomogeneities that arise from slight variations in local environments,typically associated with electric field fluctuations [9]. Whereas 1D (i.e. FTIR) spectra average over this ensemble,2D-IR spectroscopy spreads the information along the frequency diagonal (i.e. Wexite = Wdetect). In dynamically evolving systems,the frequency correlation is transient,and decays as initially excited sub-ensembles stochastically sample the accessible conformations or environments. The asymmetry of the 2D line shape can be related to the frequency fluctuation correlation function (FFCF): C(t)=<δW(0) δW(t)>,where δW(t)=W(t)-<w> is the instantaneous frequency fluctuation [10]. The decay of the FFCF arises from an equilibrium random walk through the spectral band,and is referred to as ‘‘spectral diffusion.’’ In several examples we have found that the time scale for spectral diffusion reveals the dynamics of the surrounding solvent [11, 12, 13, 14]. In our studies of biomolecular hydration,the spectral diffusion time scale is the central observable.

|
Download:
|
| Fig. 1. (A) Pulse sequence for 2D-IR spectroscopy. (B) X-ray structure of HEWL labeled with a ruthenium dicarbonyl probe. (C) Early waiting-time 2D-IR spectrum showing frequency correlation due to inhomogeneous broadening. (D) FT-IR spectrum of HEWL-RC in D2O. (E) FFCF decays for small molecules CORM-2 and PI-CORM in D2O as well as HEWL-RC. (F) Structures of CORM-2 and PI-CORM. Portions of this figure are adapted from Ref. [6]. | |
{kind=link}
Using hen egg white lysozyme (HEWL) as a model protein,we studied the ultrafast dynamics of a ruthenium probe bound to a solvent-exposed histidine (Fig. 1B),with site-specificity determined by the X-ray crystal structure [3, 4, 6]. In dilute solution of ruthenium carbonyl labeled HEWL (HEWL-RC),we find the relaxation to be well described by a fast exponential decay plus an offset. The offset is due to dynamics on time scales slower than our measurement,which is limited to~10 ps due to the <4 ps vibrational lifetime of the carbonyl modes in D2O [8]. We take the exponential time constant as a measure of the hydration dynamics, and the static offset value becomes a measure of the protein dynamics. A larger offset correlates with slower protein dynamics preventing the probe from sampling the full range of conformations and local environments.
From the HEWL measurements,we conclude that the hydration slowdown at the protein interface (2.7±0.4 ps) is roughly a factor of two compared to bulk water (1.5±0.4 ps) [4]. We and others have shown that organometallic vibrational probes reliably report the dynamics of water,and the timescales agree quantitatively with direct 2D-IR measurements of water itself (both H2O and D2O) [2, 4, 15]. The twofold slowdown is consistent with 17O magnetic relaxation dispersion measurements [16],but differs substantially from estimates using fluorescence probes [17, 18],suggesting the fluorescence approach might be more sensitive to the slower more global protein dynamics contained in our measured offsets. The twofold slowdown has been observed in explicit solvent molecular dynamics simulations by Laage et al. on the same lysozyme system [19] as well as other globular proteins [19, 20]. Extending our investigations of biomacromolecular hydration using bicelles as a model lipid bilayer [2],we also observe a 2-3-fold dynamical slowdown,suggesting that water is perturbed similarly near membranes despite the very different chemical and structural composition. Taken together,these results suggest that there is a general,modest hydration slowdown near extended biomolecular surfaces.
To assess the degree of hydration water’s coupling to the bulk, we altered the lysozyme solution by adding glycerol,a well known viscogen that preserves protein hydration [21]. Glycerol does not significantly associate with the protein surface directly; lysozyme is even functional in 99% glycerol solutions. With increased glycerol concentration,we find a monotonic,but sublinear dependence of the spectral diffusion time on the glycerol concentration [4]. The absolute change in the hydration dynamics time scale is rather weakly dependent on bulk solvent viscosity: a 100-fold increase in viscosity produces only a 2-3-fold additional slowdown in hydration dynamics. The exponential time constants and the protein-related offsets are strongly correlated,suggesting a microscopic picture of the so-called ‘‘solvent slaving’’ model of protein dynamics. That is,the hydration and protein dynamics are closely coupled,but neither is strongly influenced by the dynamics of the bulk solution. This result suggests a distance-dependent dynamic coupling between the local protein/hydration dynamics and the motional dynamics of the distant solvent.
We next turned our attention to the effects of macromolecular crowding on protein and hydration dynamics. Cells are highly crowded spaces,with macromolecule concentrations of typically 300 mg/mL [22]. Macromolecular crowders exclude volume and restrict flexibility. Favorable interactions with crowders,however, may favor unfolded or nonnative structures. Hence,in assessing thermodynamic stability it is important to vary the nature of the crowder to test the relative importance of physical (excluded volume) effects and specific chemical interactions [23].
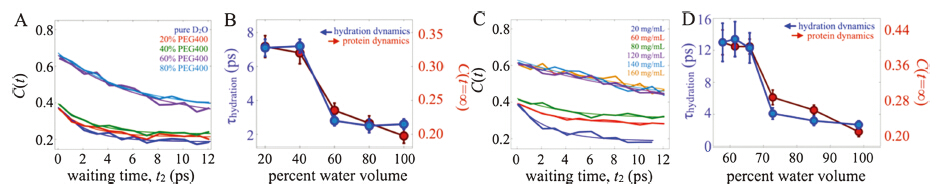
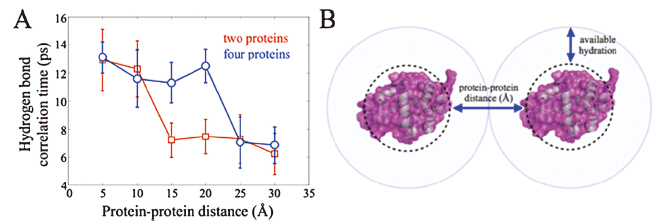
We added to the HEWL-RC solution varying amounts of either polyethylene glycol (PEG) with molecular weight of 400 Da (i.e. PEG400),or lysozyme itself. Since increasing the concentration of polymer or protein increases the solution’s viscosity,we would expect a general slowdown of the protein and hydration dynamics as we found in the glycerol study. Instead,we observed a qualitatively different trend (Fig. 2) [6]. At low crowder concentration,we saw relatively little change in hydration dynamics as reported by the vibrational label’s spectral diffusion. In contrast to the gradual slowdown with increased glycerol,at a critical macromolecular crowder concentration we observe an sharp dynamical change,which plateaus at still higher crowder concentrations. Both the PEG and lysozyme crowders induced the same type of dynamical transition,though at different crowding fractions. The transition appears at lower crowding fraction (~30%) with lyzosyme than with PEG400 (~50%),most likely due to the much richer electrostatic footprint of the highly charged (pI = 11) lysozyme. Nevertheless,the dynamical transition occurs for both cases,suggesting a physical origin which we attribute to the hydration dynamics slowdown due to the collective influence of multiple extended interfaces. With the lysozyme crowders,we can estimate the transition to occur at an interprotein separation of ~40Å ,which is similar to conclusions drawn from THz absorption spectra of crowded proteins by Havenith et al. [24, 25] The experiments are supported by explicit solvent,multi-protein MD simulations,where we analyzed the hydrogen bond correlation time as a measure of water reorientational motion. By simulating several interprotein distances,the water molecules near the protein surface were found to slow significantly at a critical protein-protein separation depending on the crowding geometry (Fig. 3). Thus,the simulations support our interpretation of a collective,cooperative hydration slowdown.

|
Download:
|
| Fig. 2. (A) FFCF decays for HEWL-RC with various concentrations (%v/v) of PEG400. (B) Protein offsets and hydration timescales extracted from fits of the FFCFs. (C) FFCFs for HEWL-RC crowded with lysozyme,and the corresponding fit parameters. Both experiments show a similar dynamical transition. Figure adapted from Ref. [4]. | |
{kind=link}

|
Download:
|
| Fig. 3. (A) Hydrogen bond correlation times determined from explicit solvent MD simulations of HEWL in one- and four-protein configurations. (B) Cartoon illustrating collective hydration due to multiple extended interfaces. Figure adapted from Ref. [4]. | |
{kind=link}
Living cells are crowded environments where interfaces between macromolecules,organelles and other large scale structures present challenges to our understanding of biomolecular function. The studies reviewed here aim to build up a systematic picture of the complex interplay between proteins and their hydration environments become altered by dynamical and structural influences. In the future,it may even be possible to perform these studies in real cells by combining 2D-IR spectroscopy with microscopic imaging [26]. Our work suggests that metal carbonyl probes are sufficiently sensitive to yield detectable signals at cell-relevant concentrations (~50 μmol/L),and probes can be incorporated either by transmembrane diffusion or by targeting using peptides. Such measurements would enable direct investigations of microscopic dynamics underlying transport in cells [27],which may help to resolve open questions regarding the importance and origins of anomalous diffusion and nonequilibrium hydrodynamics and how these influence folding and transport [28].
AcknowledgmentsThis work has been supported by the National Science Foundation (No. CHE-0748501),the National Institutes of Health (No. RR012255),and by the Camille & Henry Dreyfus Foundation.
| [1] | D. Chandler, Interfaces and the driving force of hydrophobic assembly, Nature 437 (2005) 640-647. |
| [2] | D.G. Osborne, J.A. Dunbar, J.G. Lapping, A.M. White, K.J. Kubarych, Site-specific measurements of lipid membrane interfacial water dynamics with multidimensional infrared spectroscopy, J. Phys. Chem. B 117 (2013) 15407-15414. |
| [3] | J.T. King, E.J. Arthur, C.L. Brooks, K.J. Kubarych, Site-specific hydration dynamics of globular proteins and the role of constrained water in solvent exchange with amphiphilic cosolvents, J. Phys. Chem. B 116 (2012) 5604-5611. |
| [4] | J.T. King, K.J. Kubarych, Site-specific coupling of hydration water and protein flexibility studied in solution with ultrafast 2D-IR spectroscopy, J. Am. Chem. Soc. 134 (2012) 18705-18712. |
| [5] | E.J. Arthur, J.T. King, K.J. Kubarych, C.L. Brooks, Heterogeneous preferential solvation of water and trifluoroethanol in homologous lysozymes, J. Phys. Chem. B 118 (2014) 8118-8127. |
| [6] | J.T. King, E.J. Arthur, C.L. Brooks, K.J. Kubarych, Crowding induced collective hydration of biological macromolecules over extended distances, J. Am. Chem. Soc. 136 (2014) 188-194. |
| [7] | P. Ball, Water as an active constituent in cell biology, Chem. Rev. 108 (2008) 74- 108. |
| [8] | J.T. King, M.R. Ross, K.J. Kubarych, Water-assisted vibrational relaxation of a metal carbonyl complex studied with ultrafast 2D-IR, J. Phys. Chem. B 116 (2012) 3754- 3759. |
| [9] | P. Hamm, M.T. Zanni, Concepts and Methods of 2D Infrared Spectroscopy, Cambridge University Press, New York, 2011. |
| [10] | S. Roberts, J. Loparo, A. Tokmakoff, Characterization of spectral diffusion from two-dimensional line shapes, J. Chem. Phys. 125 (2006) 084502. |
| [11] | D.G. Osborne, J.T. King, J.A. Dunbar, A.M. White, K.J. Kubarych, Ultrafast 2DIR probe of a host-guest inclusion complex: structural and dynamical constraints of nanoconfinement, J. Chem. Phys. 138 (2013) 144501. |
| [12] | D.G. Osborne, K.J. Kubarych, Rapid and accurate measurement of the frequency- frequency correlation function, J. Phys. Chem. A 117 (2012) 5891-5898. |
| [13] | J.T. King, M.R. Ross, K.J. Kubarych, Ultrafast alpha-like relaxation of a fragile glassforming liquid measured using two-dimensional infrared spectroscopy, Phys. Rev. Lett. 108 (2012) 157401. |
| [14] | J.T. King, C.R. Baiz, K.J. Kubarych, Solvent-dependent spectral diffusion in a hydrogen bonded ‘‘Vibrational Aggregate'', J. Phys. Chem. A 114 (2010) 10590- 10604. |
| [15] | J.F. Brookes, K.M. Slenkamp, M.S. Lynch, M. Khalil, Effect of solvent polarity on the vibrational dephasing dynamics of the nitrosyl stretch in an FeII complex revealed by 2D IR spectroscopy, J. Phys. Chem. A 117 (2013) 6234-6243. |
| [16] | J. Qvist, E. Persson, C. Mattea, B. Halle, Time scales of water dynamics at biological interfaces: peptides, proteins and cells, Faraday Discuss. 141 (2009) 131-144. |
| [17] | W.H. Qiu, Y.T. Kao, L.Y. Zhang, et al., Protein surface hydration mapped by sitespecific mutations, Proc. Natl. Acad. Sci. U. S. A. 103 (2006) 13979-13984. |
| [18] | S.K. Pal, J. Peon, A.H. Zewail, Ultrafast surface hydration dynamics and expression of protein functionality: alpha-chymotrypsin, Proc. Natl. Acad. Sci. U. S. A. 99 (2002) 15297-15302. |
| [19] | F. Sterpone, G. Stirnemann, D. Laage, Magnitude and molecular origin of water slowdown next to a protein, J. Am. Chem. Soc. 134 (2012) 4116-4119. |
| [20] | A.C. Fogarty, D. Laage, Water dynamics in protein hydration shells: the molecular origins of the dynamical perturbation, J. Phys. Chem. B 118 (2014) 7715-7729. |
| [21] | T. Knubovets, J.J. Osterhout, P.J. Connolly, A.M. Klibanov, Structure, thermostability, and conformational flexibility of hen egg-white lysozyme dissolved in glycerol, Proc. Natl. Acad. Sci. U. S. A. 96 (1999) 1262-1267. |
| [22] | A.P. Minton, The influence of macromolecular crowding and macromolecular confinement on biochemical reactions in physiological media, J. Biol. Chem. 276 (2001) 10577-10580. |
| [23] | M. Sarkar, J. Lu, G.J. Pielak, Protein crowder charge and protein stability, Biochemistry 53 (2014) 1601-1606. |
| [24] | S. Ebbinghaus, S.J. Kim, M. Heyden, et al., An extended dynamical hydration shell around proteins, Proc. Natl. Acad. Sci. U. S. A. 104 (2007) 20749-20752. |
| [25] | V.C. Nibali, M. Havenith, New insights into the role of water in biological function: studying solvated biomolecules using terahertz absorption spectroscopy in conjunction with molecular dynamics simulations, J. Am. Chem. Soc. 136 (2014) 12800-12807. |
| [26] | C.R. Baiz, D. Schach, A. Tokmakoff, Ultrafast 2D IR microscopy, Opt. Express 22 (2014) 18724-18735. |
| [27] | T. Ando, J. Skolnick, Crowding and hydrodynamic interactions likely dominate in vivo macromolecular motion, Proc. Natl. Acad. Sci. U. S. A. 107 (2010) 18457- 18462. |
| [28] | A. Gershenson, L.M. Gierasch, Protein folding in the cell: challenges and progress, Curr. Opin. Struct. Biol. 21 (2011) 32-41. |