




Dopexamine dihydrochloride (1) was originally developed by Fisons Corporation [1]. It was first marketed in Ireland in 1989 and was not yet in Chinese market. As a dopamine (DA) analogue, dopexamine is a DA receptor agonist and β2-adrenoceptor agonist, with weak β2-adrenergic activity and no activity at α-adrenoceptors [2, 3]. It is commonly used in clinical treatment of acute heart failure [4],as well as low cardiac output after cardiac surgery [5], septic shock [6],etc.
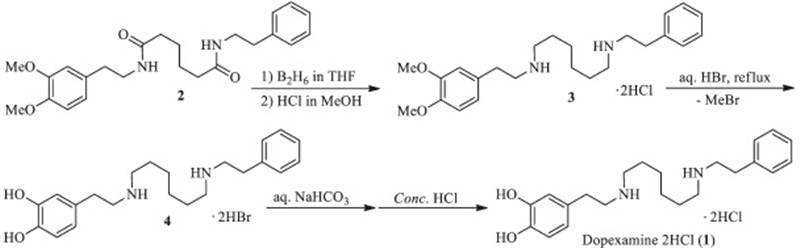
Several synthetic routes of the target have been reported. Fisons Corporation [1] developed the synthesis from the diborane reduction of the di-amide 2 which could be easily prepared from the condensation of 6-((3,4-dimethoxyphenethyl)amino)-6-oxohexanoic acid and 2-phenylethylamine. Then the di-amine 3 was demethylated to provide dopexamine in dihydrobromic form (6). After release of the salt to its free base,itwas finally converted to the required dihydrochloride salt (1) (Scheme 1). The total yield from the hexanoic acid was 37.5%. Lu and co-workers [7] described two alternative methods for the synthesis of the target,where the formation of the amides was performed harshly under molten condition and side reactions might occur in the process in under 25% overall yield. In all the above routes,methyl group was employed to protect phenolic hydroxyl group. Nevertheless,the deprotection had to be carried out in strong aqueous hydrobromic acid under refluxing,which led to the formation of highly toxicCH3Br and some other by-products. It has to be mentioned that the bromide anion is not acceptable due to the injection formulation of the drug,so it is critical to free the base from the salt 6 via alkalization and to wash with water to completely remove the bromide. In the operation, the base might be easily oxidized due to the typical catecholamine structure. Finally,the base was converted to the required dihydrochloride salt.

|
Download:
|
| Scheme 1.The Fison’s synthetic route of dopexamine 2HCl via Me-protection. | |
{kind=link}
In order to obtain the medical-grade and high-purity target,and to minimize the types and the contents of possible impurities,it is essential to achieve an optimal improvement of the above syntheses. In this paper,the tuning of protecting group was a key consideration,which should be in good tolerance and could be deprotected under mild condition.
Based on these considerations,here is developed a novel strategy for the synthesis of the product. Instead of methyl protecting method,benzyl group was first employed for the protection of the phenolic hydroxyl in the strategy. This protecting group could not only effectively protect the phenol,but also be mildly removed via catalytic hydrogenation in hydrochloric solvent at room temperature. This method was able to concisely get the hydrochloride of dopexamine. It avoided all disadvantages when methyl group was used,such as the toxicCH3Br formation,the release the hydrobromic salt and re-formation of the hydrochloric salt. 2. Experimental
All solvents were dried and distilled before use,and all reagents were procured from commercial sources and used without further purification. Silica gel GF254 and silica gel (200-300 mesh) were respectively used for thin-layer chromatography and column chromatography. Melting point data were determined by X-4 micromelting point apparatus (Beijing Tech Instrument Co.) and uncorrected. Mass spectra were performed by a Finnigan LCQ LS/ MS instrument,1H NMR and 13C NMR spectra were recorded by a Bruker ARX-300 or ARX-600 spectrometer with TMS as the internal standard. High Performance Liquid Chromatography (HPLC) was carried out with a Shimadzu LC-2010CHT. The residual palladium was determined by ICP-MS (XSeries II).
The purity of the final compound 1 was analyzed by HPLC under the following conditions: column,Inertsil ODS-SP,5 μm, 150 mm×4.6 mm; flow rate,1.0mL/min; detection at 280 nm and 210 nm; mobile phase,solution A: mix 5 volumes of buffer solution pH 2.5 (dissolve 100 g of potassium dihydrogen orthophosphate in 800mL of water,adjust to pH 2.5 with hydrochloric acid and add sufficient water to produce 1000 mL) and 95 volumes of water; solutionB:mix5 volumes of buffer solutionpH2.5 and 95 volumes of 60% (v/v) solution of acetonitrile; gradient elution according to BP2013(BritishPharmacopoeia2013);Retentiontimefor compound 1 was about 7.5 min.
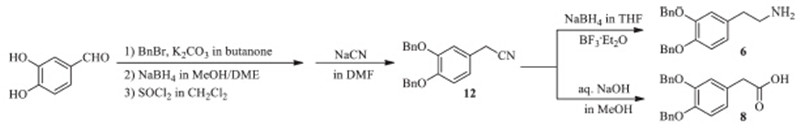
2-(3,4-Bis(benzyloxy)phenyl)ethylamine (6) (Scheme 2): It was synthesized from the acetonitrile 12,which was afforded from 3,4- dihydroxybenzaldehyde in 70.5% over four steps (Scheme 3). To a solution of 12(65.9 g,0.2 mol) in 500 mL THF (tetrahydrofuran), NaBH4 (37.8 g,1.0 mol) was added,then BH3-Et2O (25.6 mL, 1.0 mol) was dropwise added at room temperature. After addition, the reaction mixture was heated under reflux for 12 h. Then the solid material was filtered off and the filtrate was concentrated under vacuum. The residue was dissolved in the mixture of ice water (500 mL) and ethyl acetate (300 mL). Then the organic phase was separated,and the aqueous residue was extracted with ethyl acetate (2 ×100 mL). The combined organic phase was washed with water (2 × 200 mL),dried over anhydrous Na2SO4,filtered and the filtrate was concentrated under vacuum to afford 6 as light yellow oil (59.0 g,88.5%). MS (ESI) m/z: 333.9 [M+H]+.

|
Download:
|
| Scheme 2.The synthesis of two key intermediates 6 and 8. | |
{kind=link}

|
Download:
|
| Scheme 3.Our concise synthesis of dopexamine 2HCl via Bn-protecting protocol. | |
{kind=link}
2-(3,4-Bis(benzyloxy)phenyl)acetic acid (8) (Scheme 2): It could also be prepared from the acetonitrile 12 (Scheme 3). To a solution of sodium hydroxide (24.0 g,0.6 mol),water (100 mL) and MeOH (200 mL),12 (32.9 g,0.1 mol) was added. The mixture was heated under reflux for 6 h,and then concentrated under vacuum to distill off MeOH. The aqueous residue was dissolved in the mixture of ice water (200 mL) and ethyl acetate (200 mL). After acidification with dil. HCl (pH 2-3),the organic phase was separated,and the aqueous residue was extracted with ethyl acetate (2 ×50 mL). The combined organic phase was washed with water (2 ×200 mL),dried over anhydrous Na2SO4,filtered and the filtrate was concentrated under vacuum to afford 8 as light yellow solid (32.5 g,93.4%),m.p. 107-108 ℃. MS (ESI)m/z: 371.4 [M+Na]+, 346.8 [M-H]-.
The di-amide 7—N-(3,4-bis(benzyloxy)phenethyl)-6-(2-phenylacetamido) hexanamide [1]: It was prepared from the ethylamine 6 and 6-(2-phenylacetamido)hexanoic acid (5) which was synthesized with 2-phenylacetic acid and 6-amino hexanoic acid in 81.1% (Scheme 3). To a solution of 5 (2.0 g,8.0 mmol) in DCM (50 mL),CDI (1.95 g,12.0 mmol) was added. After stirring for 2 h at room temperature,6(2.8 g,8.0 mmol) was added. After stirring for another 10 h,the reaction mixture was poured into ice water (100 mL),the organic phase was separated,and the aqueous residue was extracted with DCM (2× 20 mL). The combined organic phase was washed with water (2 × 50 mL),dried over anhydrous Na2SO4,filtered and the filtrate was concentrated under vacuum. The residue was purified by silica gel column chromatography to afford 7 as white solid (3.84 g,81.9%). 1H NMR (CDCl3, 300 MHz): δ 1.18-1.26 (m,2H),1.38-1.43 (m,2H),1.52-1.57 (m, 2H),2.04 (t,2H,J = 7.5 Hz),2.69 (t,2H,J = 6.9 Hz),3.15-3.19 (dd, 2H,J = 13.3,13.3 Hz),3.40-3.44 (t,2H,J = 6.6 Hz),3.53 (s,2H),5.13 (s,4H),6.70-6.89 (m,3H),7.24-7.45 (m,15H).
Preparation of 9: (1) To a cooled solution (below 5 ℃) of 8 (24.4 g,70.0 mmol) and N-hydroxysuccinimide (9.7 g,84.0 mmol) in DCM (300 mL),N,N-dimethylaniline (0.4 g,3.5 mmol) and dicyclohexyl carbodiimide(17.3 g,84.0 mmol) were successively added. After addition,the mixture was stirred for 24 h at room temperature. The solid material was filtered off and the filtrate was concentrated under vacuum to afford the active ester which was used for the next step without further purification. (2) To a cooled mixture (below 10 ℃) of 6-amino hexanoic acid (18.3 g, 140 mmol),Et3N (24.4 mL,175 mmol),water (150 mL) and acetonitrile (50 mL),the solution of the active ester in acetonitrile (100 mL) was dropwisely added. After addition,the mixture was stirred for 3 h at room temperature,and then concentrated under vacuum. The residue was dissolved in the mixture of ice water (200 mL) and ethyl acetate (200 mL). After acidification with dil. HCl (pH 2-3),the organic phase was separated,and the aqueous residue was extracted with ethyl acetate (2 × 50 mL). The combined organic phase was washed with water (2 ×100 mL), dried over anhydrous Na2SO4,filtered and the filtrate was concentrated under vacuum. The residue was purified by silica gel column chromatography to afford 9 as light yellow solid (29.8 g,92.3%),m.p. 108-110 ℃. MS (ESI) m/z: 462.1 [M+H]+, 484.4 [M+Na]+,460.2 [M-H]-,1H NMR (CDCl3,400 MHz): δ 1.25- 1.27 (m,2H),1.36-1.38 (m,2H),1.57-1.60 (m,2H),2.28-2.32 (m, 2H),3.13-3.15 (m,2H),3.450 (s,2H),5.15 (s,4H),4.40-5.42 (m, 1H),6.74-6.92 (m,3H),7.29-7.45 (m,10H),13C NMR (600 MHz, CDCl3): δ 24.21,26.11,28.99,33.78,39.40,43.17,71.22,71.31, 115.41,116.28,122.53,127.32,127.36,127.88,128.51,136.99, 137.13,148.32,149.10,171.68,178.15.
The di-amide 10—6-(2-(3,4-bis(benzyloxy)phenyl)acetamido)- N-phenethylhexanamide [1]: To the solution of 9 (8.3 g, 18.0 mmol) in DCM (200 mL),CDI (5.8 g,27.0 mmol) was added. After stirring for 2 h at room temperature,2-phenyl ethanamine (4.5 mL,27.0 mmol) was added. After stirring for another 5 h,the reaction mixture was poured into ice water (300 mL),the organic phase was separated,and the aqueous residue was extracted with DCM (2 × 50 mL). The combined organic phase was washed with water (2 × 100 mL),dried over anhydrous Na2SO4,filtered and the filtrate was concentrated under vacuum. The residue was purified by silica gel column chromatography to afford 10 as white solid (8.8 g,86.3%) (Scheme 2). MS (ESI) m/z: 565.3 [M+H]+, 563.1 [M-H]-. 1H NMR (CDCl3,600 MHz): δ 1.19-1.21 (m,2H), 1.36-1.38 (m,2H),1.54-1.56 (m,2H),2.06 (t,2H,J = 7.8 Hz),2.78 (t,2H,J = 7.2 Hz),3.11-3.15 (dd,2H,J = 6.6,7.2 Hz),3.44 (s,2H), 3.46-3.50 (dd,2H,J = 13.2,13.8 Hz),5.14 (s,4H),6.74-6.91 (m, 3H),7.16-7.43 (m,15H).
The di-amine 11—N1-(3,4-bis(benzyloxy)phenethyl)-N6-phenethylhexane- 1,6-diamine: To a cooled solution (below 20 ℃) of 7 or 10 (14.1 g,25.0 mmol) in THF (150 mL),NaBH4 (5.7 g, 150.0 mmol) was added. Then acetic acid (8.6 mL)was dropwisely added. After addition,the reaction mixture was heated under reflux for 12 h. The solid material was filtered off and the filtrate was concentrated under vacuum. The residuewas dissolved in the mixture of ice water (200 mL) and ethyl acetate (100 mL). Then the organic phase was separated,and the aqueous residue was extracted with ethyl acetate (2 × 50 mL). The combined organic phase was washed with water (2 × 50 mL),dried over anhydrous Na2SO4,filtered and the filtrate was concentrated under vacuum. The obtained amine was treated with the solution of HCl in MeOH under reflux for 30 min,then the mixture was cooled and filtered to afford 11 as white solid (from 7: 13.6 g,89.2%,from 10: 14.0 g, 91.9%) (Scheme 2). MS (ESI) m/z: 537.3 [M+H]+. 1H NMR (DMSOd6, 300 MHz): δ 1.34 (s,4H),1.63 (s,4H),2.86-3.23 (m,12H),5.11 (d,4H,J = 5.3 Hz),6.76-7.02 (m,3H),7.26-7.47 (m,15H),8.90 (br,4H).
Dopexamine dihydrochloride (1) (Scheme 3): To the solution of 11 (10.0 g) in MeOH (200 mL),Pd/C (10%,1.0 g) was added,the mixture was stirred under hydrogen atmosphere for 10 h. Then the mixture was filtered and the filtrate was concentrated to afford crude product 6.9 g. The solid was recrystallized from MeOH to afford 1 as white solid (5.8 g,87.1%),m.p. 218-220 ℃. MS (ESI) m/z: 357.2 [M+H]+. 1H NMR (DMSO-d6,300 MHz): δ 1.32 (s,4H), 1.62 (s,4H),2.74-3.15 (m,12H),6.45-6.68 (m,3H),7.21-7.25 (m,5H),8.84-9.10 (m,6H); 13C NMR (DMSO-d6,75 MHz): δ 25.26, 25.54,31.00,31.55,46.56,47.80,48.25,115.86,116.11,119.25, 126.81,127.97,128.70,137.44,144.15,145.41. 3. Results and discussion
In the structure of the target,two secondary amines was connected with the left dopamine,the right phenylethylamine,and the between six-carbon-unit linker. Thus,the left part could come from the di-Bn-protected dopamine 6 or the acetic acid 8, and the v-aminocaproic acid was selected as the building block for the six-carbon-unit linker. The Bn-protected di-amine 11 could be obtained from the reduction of the corresponding di-amide 7 or 10.
Accordingly,two synthetic routes were studied in this paper. In Route I,after the condensation of phenylacetic acid with ω-aminocaproic acid,the resulting carboxylic acid 5 was reacted with 2-(3,4-bis(benzyloxy)phenyl)ethylamine (6) for the formation of the diamide 7 (Scheme 3). In Route II,2-(3,4-bis(benzyloxy)phenyl) acetic acid (8) was first condensed with ω-aminocaproic acid to form the acid 9 which was then condensed with 2-phenylethanamine to form the second amide 10. The resulting di-amides (7 or 10) from both routes were reduced with NaBH4 to afford the di-amine 11. Finally,the key intermediate 11 was deprotected via catalytic hydrogenation in hydrochloric methanol at room temperature to directly afford the final product in dihydrochloric salt. The overall yields were 43.8% and 54.1% for Route I from phenylacetic acid and for Route Ⅱ from 2-(3,4-bis(benzyloxy)phenyl) acetic acid (8),respectively. The improved strategy simplified the procedure and avoided all disadvantages when methyl group was used.
By the way,2-(3,4-bis(benzyloxy)phenyl)ethylamine 6 and 2- (3,4-bis(benzyloxy)phenyl)acetic acid 8 could be easily prepared from the same precursor 2-(3,4-bis(benzyloxy)phenyl)acetonitrile (12) which derived from commercial available 3,4-dihydroxybenzaldehyde. The reduction of 12 provided 6 while hydrolysis of it afforded 8. 4. Conclusion
Benzyl protecting protocol was first employed in two routes for the concise synthesis of dopexamine dihydrochloride. This protecting group could be cleanly removed under mild condition and no unacceptable ion was brought to the final product. The total yield of Route I was 43.8% from phenylacetic acid,while it was 54.1% of Route II from 2-(3,4-bis(benzyloxy)phenyl)acetic acid. The titration purity of the final product was more than 99.5%,while any single or total impurities met the known standard of the drug by HPLC analysis. The measured residual palladium (63.20 ppb) met an acceptable limit of the metal (<1 ppm) as an API for injection. Therefore,the developed synthetic routes will be benefit in the industrial production of dopexamine dihydrochloride.
Acknowledgments
This project was supported by Program for Innovative Research Team of the Ministry of Education and Program for Liaoning Innovative Research Team in University. Financial support was generously given from Shenyang Shuangding Pharmaceutical Co., LTD.,Shenyang,China.
| [1] | O. Olejnik, Pharmaceutical formulations, EP0117033A2, 1984. |
| [2] | T. Meinertz, H. Drexler, H. Just, Dopexamine in congestive heart failure: how do the pharmacological activities translate into the clinical situation? Basic Res. Cardiol. 84 (1989) 177-186. |
| [3] | R.A. Brown, J. Dixon, J.B. Farmer, et al., Dopexamine: a novel agonist at peripheral dopamine receptors and b2-adrenoceptors, Br. J. Pharm. 85 (1985) 599-608. |
| [4] | C.V. Leier, P.F. Binkley, J. Carpenter, et al., Cardiovascular pharmacology of dopexamine in low output congestive heart failure, Am. J. Cardiol. 62 (1988) 94-99. |
| [5] | B.D. Gres, C. Flamens, M.C. Gruner, The use of dopexamine hydrochloride in patients with low cardiac output after cardiac surgery, J. Cardiothorac. Anest. 3 (1989) 16-19. |
| [6] | F.C. Colardyn, J.F. Vandenbogaerde, D.P. Vogelaers, et al., Use of dopexamine hydrochloride in patients with septic shock, Crit. Care Med. 17 (1989) 999-1003. |
| [7] | J. Lu, Y. Ma, L. Mo, et al., Synthesis of dopexamine, J. China Pharm. Univ. 30 (1999) 328-331. |