




b Guizhou University, Guiyang 550002, China
Carbazole is a significant heterocyclic template broadly found in many natural products. Compounds containing carbazole core structures are of great pharmaceutical interest due to their potent biological activity and medicinal significance. Consequently,many synthetic protocols have been developed to access these frameworks [1].
Furostifoline (Fig. 1),the first furocarbazole alkaloid obtained from natural sources,was isolated from the root bark ofMurraya euchrestifolia[2]. The extracts of the leaves and bark of this shrub growing in Taiwan have been used in folk medicine. Several years later,another furo[3, 2, a]carbazole alkaloid furoclausine-A has been isolated from the root bark ofClausena excavate[3]. Extracts of this plant have been used for the treatment of various infections and poisonous snakebites in traditional Chinese folk medicine. Furostifoline and furoclausine-A both contain a furo[3, 2, a]carbazole core and represent a relatively young class of alkaloids. Even though the structures of these natural products are relatively simple,their pharmacological potential and structure features made them became attractive synthetic targets. Interest in these alkaloids has resulted in at least seven total syntheses by using a variety of ways involving iron-mediated method,photocyclization methodologies [4] and rhodium(II)-catalyzed cyclization of bis(N-tosylhydrazone)s [5]. Moreover,de Koning and co-workers [6] applied the Wittig/RCM process to construct the central aromatic ring of both the indolo[2, 3, a]carbazole and the thieno[3, 2, a]carbazole nucleus,however,using the same methodology for synthesis of furostifoline was unsuccessful.

{kind=link}
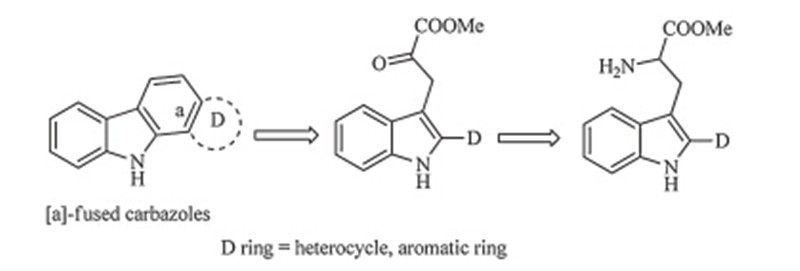
It was well known thata-ketocarbonyl compounds are found as metabolic intermediates in the biosynthesis of amino acids and generated by a cofactor-mediated transaminase reaction. This biosynthetic approach,due to its potential benefits for organic synthesis,has garnered significant attention from the chemical community. Conditions for non-enzymatic transamination were developed [7, 8] and played an important role in the synthesis of natural products [9, 10]. Meanwhile,although indole 3-pyruvic acid and its derivatives are key intermediates in the biosynthetic route of several indole alkaloids such as rebeccamycin [11, 12] and eusynstyelamides A-C [13]. To the best of our knowledge,these biosynthetic indications and fragment convenience were rarely involved in the process of developing synthetic strategies for carbazoles. This fact enabled us to investigate whether a transamination step based on tryptophan derivatives could be utilized in the synthesis of furo[3, 2, a]carbazole alkaloids and other [a]-fused carbazoles (Scheme 1). Intrigued by these considerations, we report a new approach to furo[3, 2, a]carbazole in this paper.

|
Download:
|
| Scheme 1.A novel synthetic strategy to [a]-fused carbazoles. | |
{kind=link}
The procedure for preparation of all intermediates in Scheme 2 is listed as below:

|
Download:
|
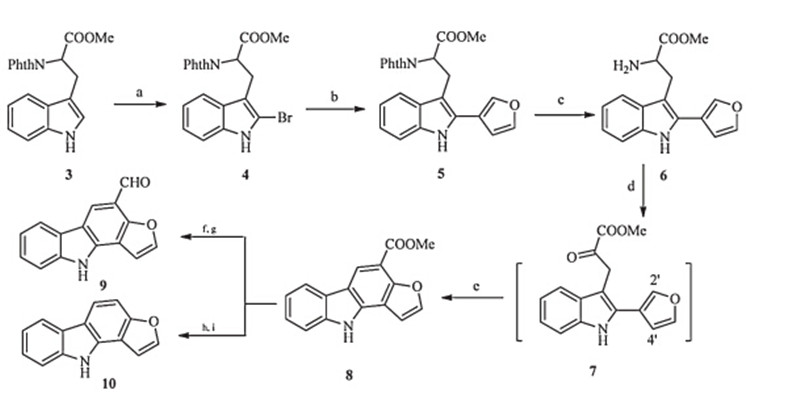
| Scheme 2.Reagents and conditions: (a) PyHBr3,THF/CHCl3,94%; (b) Pd(OAc)2,PPh3,Et3N,DMF,1008C,61%; (c) N2H4·H2O,CH2Cl2/MeOH,80%; (d) ZnSO4·7H2O, CHOCOONa·H2O,MeCN/acetate buffer; (e) TFA,1,4-dioxane,1008C,58% for two steps; (f) LiAlH4,THF; (g) Dess-Martin periodinane,CH2Cl2,87% for two steps; (h) NaOH, MeOH/H2O; (i) 3008C,63% for two steps. | |
{kind=link}
Preparation of compound 4: A solution of compound 3 (2.0 g, 5.7 mmol) in THF:CHCl3(1:1,10 mL) was stirred at 0℃ for 15 min. Pyridium tribromide (2.02 g,6.3 mmol) was then added portionwise over 1 h. Saturated aqueous Na2S2O3(25 mL) was added,and following a colour change,saturated aqueous NaHCO3(25 mL) was also added. The aqueous phase was extracted with CHCl3 (3×20 mL),the combined organic phase were dried over Na2SO4 and concentrated under reduced pressure. The crude oil was purified by column chromatography (petroleum ether: EtOAc = 3:1,v/v) to afford compound 4 as a yellow solid (2.31 g, 5.4 mmol,94%). IR (KBr,cm-1 ):n3351,1713,1390,1256,719,531; 1H NMR (400 MHz,CDCl3):δ3.63-3.70 (m,2H),3.80 (s,3H),5.20 (dd,1H,J= 10.4,5.4 Hz),6.94-6.98 (m,1H),7.02-7.06 (m,1H), 7.13-7.16 (m,1H),7.45 (d,1H,J= 7.6 Hz),7.64 (dd,2H,J= 5.6, 2.8 Hz),7.73 (dd,2H,J= 5.6,2.8 Hz),8.11 (br,1H); 13C NMR (100 MHz,CDCl3):δ169.4,167.4,135.8,134.0,131.6,127.5,123.3, 122.3,120.1,117.6,110.5,109.1,52.9,51.7,24.6; ESI-MS:m/z 427 [M+H]+ ,449 [M+Na]+ .
Preparation of compound 5: A solution of compound 4(1.0 g, 2.34 mmol),furan-3-boronic acid (472 mg,4.2 mmol),Et3N (976mL,7.02 mmol),PPh3 (122 mg,0.47 mmol),and Pd(OAc)2 (53 mg,0.23 mmol) in DMF (5 mL) was stirred at 100℃ for 10 h under nitrogen atmosphere. After cooling to room temperature, the mixture was quenched with water,and then the mixture was extracted with EtOAc. The EtOAc layer was washed with water and brine,dried over Na2SO4 and concentrated. The residue was purified by column chromatography (petroleum ether: EtOAc = 5:1,v/v) to afford compound 5 as a yellow solid (582 mg,1.36 mmol,61%). IR (KBr,cm-1 ): v3380,1773,1711, 1391,873,719,597,530; 1H NMR (500 MHz,CDCl3):δ3.74-3.86 (m,2H),3.78 (s,3H),5.10 (dd,1H,J= 10.6,4.6 Hz),6.54 (s,1H),6.98 (t,1H,J= 7.4 Hz),7.08 (t,1H,J= 7.4 Hz),7.23 (d,1H,J= 8.1 Hz),7.38 (t,1H,J= 1.6 Hz),7.51 (d,1H,J= 7.9 Hz),7.61-7.64 (m,3H),7.65- 7.68 (m,2H),7.91 (br,1H); 13C NMR (125 MHz,CDCl3):δ169.5, 167.3,143.6,139.6,135.5,133.8,131.6,128.8,127.7,123.2,122.2, 119.8,118.2,117.7,110.6,109.2,108.2,52.8,52.3,24.3; ESI-MS: m/z437 [M+Na]+ ,851 [2M+Na]+ .
Preparation of compound 6: Compound 5(400 mg,0.97 mmol) was dissolved in CH2Cl2/MeOH (1:1,6 mL),80% hydrazine hydrate (0.23 mL,3.86 mmol) was added,and then the mixture was stirred at room temperature overnight. The precipitate was filtered and the filtrate was concentrated. The residue was purified by column chromatography (petroleum ether: EtOAc = 3:1,v/v) to afford the compound 6 as a brown solid (220 mg,0.78 mmol,80%). IR (KBr, cm-1 ): n2920,1737,1440,1209,873,746; 1H NMR (500 MHz, CDCl3): d 3.12 (dd,1H,J= 14.4,8.8 Hz),3.38 (dd,1H,J= 14.4, 8.8 Hz),3.68 (s,3H),3.88 (dd,1H,J= 8.8,5.0 Hz),6.74 (dd,1H, J= 1.9,1.0 Hz),7.11-7.14 (m,1H),7.17-7.19 (m,1H),7.34 (d,1H, J= 8.1 Hz),7.54 (t,1H,J= 1.8 Hz),7.60 (d,1H,J= 7.9 Hz),7.89 (dd, 1H,J= 1.5,1.0 Hz),8.12 (br,1H); 13C NMR (125 MHz,CDCl3): d 175.7,143.8,139.8,135.7,129.1,128.0,122.4,119.9,118.7,117.9, 110.7,109.4,108.6,55.2,52.0,30.6; ESI-MS:m/z307 [M+Na]+ .
Preparation of compound 8: A mixture of compound 6(200 mg, 0.70 mmol),ZnSO4·7H2O (101 mg,0.35 mmol),glyoxylic acid sodium monohydrate (396 mg,3.5 mmol) in 3 mL of MeCN and 3 mL of buffer (pH 5,containing 495 mg sodium acetate and 90mL acetic acid) was stirred at room temperature for 0.5 h. Then the mixture was extracted with EtOAc (3×10 mL),the combined organic layer were dried over Na2SO4 and concentrated under reduced pressure. The crude product was dissolved in dioxane (3.0 mL),TFA (0.1 mL) added,the reaction mixture was refluxed for 5 h. After cooling,the solvents was evaporated,and the residue was purified by column chromatography (petroleum ether: EtOAc = 3:1,v/v) to afford the compound 8 as a black solid (108 mg,58%). IR (KBr,cm-1 ): n3305,2923,1684,1639,1369, 1257,1237,745; 1H NMR (400 MHz,CDCl3):δ4.07 (s,3H),7.04 (d, 1H,J= 2.4 Hz),7.26-7.35 (m,1H),7.42-7.46 (m,1H),7.51 (d,1H, J= 8.0 Hz),7.83 (d,1H,J= 2.4 Hz),8.12 (d,1H,J= 7.6 Hz),8.72 (br, 1H),8.74 (s,1H); 13C NMR (100 MHz,CDCl3):δ166.2,153.3,144.8, 139.3,135.8,125.5,123.9,120.8,120.7,120.1,117.8,112.7,111.1, 108.0,103.3,52.1; ESI-MS:m/z288 [M+Na]+ ,553 [2M+Na]+ .
Preparation of 9: Compound 8 (200 mg,0.75 mmol) was dissoved in anhydrous THF (2 mL),LiAlH4 (57 mg,1.5 mmol) was added at 0℃. The solution was then allowed to warm to ambient temperature over 1 h. After 10% Na2SO3 solution was added,the mixture was extracted with EtOAc (3×10 mL),the combined organic phase were dried over Na2SO4and concentrated under reduced pressure. Dess-Martin periodinane (239 mg, 0.75 mmol) was added to the above crude alcohol in CH2Cl2 (3 mL) at 0℃. After stirring at 0℃ for 0.5 h,the reaction mixture was diluted with ether,and then washed with 10% Na2S2O3 solution,10% Na2SO3solution and water. Organic layer was dried and evaporated,and then the residue was purified by column chromatography (petroleum ether: EtOAc = 3:1,v/v) to afford the compound 9 as a white solid (171 mg,87% for two steps). IR (KBr, cm-1 ):n3294,1672,1636,1582,1470,1234,1179,750; 1H NMR (400 MHz,DMSO-d6):δ7.26-7.30 (m,1H),7.31 (d,1H,J= 2.4 Hz), 7.42-7.46 (m,1H),7.61-7.64 (m,1H),8.19 (d,1H,J= 2.0 Hz),8.24 (d,1H,J= 7.6 Hz),8.65 (s,1H),10.29 (s,1H),12.37 (s,1H); 13C NMR (100 MHz,DMSO-d6): d188.0,152.4,145.7,139.8,136.9,125.5, 123.3,122.0,120.3,120.2,117.3,115.0,112.5,111.7,104.3; ESIMS:m/z258 [M+Na]+ .
Preparation of compound 10: The compound 8 (100 mg, 0.38 mmol) was dissolved in MeOH/H2O (1.5 mL,2:1),NaOH (76 mg,1.90 mmol) was added and the mixture was stirred at 25℃ for 20 h. The reaction was quenched by addition of 5% HCl (2 mL) and the aqueous layer was extracted with EtOAc. The combined organic layers were washed with brine,dried with Na2SO4 and the solvents were evaporated. The crude acid was heated to 300℃ for 15 min under argon,and then the mixture was purified by column chromatography (petroleum ether: EtOAc = 3:1,v/v) to afford the compound 10 as a white solid (49 mg,63% for two steps). IR (KBr,cm-1 ): n3419,2923,1635, 1432,1378,1228,1050,743,449; 1H NMR (400 MHz,CDCl3): d 7.00 (d,1H,J= 2.4 Hz),7.28-7.30 (m,1H),7.38-7.46 (m,2H),7.51 (d,1H,J= 8.0 Hz),7.72 (d,1H,J= 2.0 Hz),7.99 (d,1H,J= 8.0 Hz), 8.09 (d,1H,J= 8.0 Hz),8.37 (br,1H); 13C NMR (100 MHz,CDCl3):δ 154.9,143.9,138.8,132.5,124.5,124.0,119.8,119.7,117.8,116.6, 111.9,110.8,104.3,103.4; ESI-MS:m/z208 [M+H]+ .
1H NMR and 13C NMR spectra for compounds 4-10 are included in the Supporting information. 3. Results and discussion
Our synthesis started with the selective bromination ofNphthaloyl(Phth) tryptophan methyl ester3at the 2-position with pyridine hydrobromide perbromide [14]. Further Suzuki coupling was realized by the adoption of triethylamine and Pd(PPh3)4in DMF at 100℃ effectively cross-coupled4with furan-3-boronic acid in 61% yield. This resulting product5was then treated with hydrazine hydrate to remove Phth group and provided the amine6 in 80% yield.
With the free amine 6 in hand,next step was set for the key transamination. After examine several reported conditions [7, 8], we found the original condition using glyoxylate as an electrophile and copper(II) ions as catalysts was not work well due to copper(II) ions would significantly cause undesired decomposition of intermediate7in the presence of oxygen or air. However,we were able to circumvent this shortage by discovering zinc sulfate as a more suitable metal catalyst for substrate 6,the reaction proceeded smoothly. Since decomposition was concomitant during the course of purification,crude intermediate 7 was directly subjected to TFA promoted aromatization in heated dioxane,the expected cyclization occurred uneventfully,thus provided the ester 8 in good yield. To our delight,good regioselectivity was achieved in this aromatic cyclization step and the 4' -position of furan fragment has not been attacked by ketone group.
The reduction of above methyl ester by LiAlH4 gave the corresponding alcohol,it was subsequently oxidated with Dess- Martin periodinane to furnish aldehyde 9 which could be viewed as dehydroxylfuroclausine-A. In a parallel synthesis,the ester was hydrolyzed,then decarboxylation of the acid intermediate happened at 300℃ [15] yielding the final product 10 which represents the basic structure features of natural furo[3, 2, a]carbazole alkaloids. 4. Conclusion
In conclusion,we have demonstrated a novel synthesis route to furo[3, 2, a]carbazole from 2-furyl tryptophan derivative. Since heteroaryl-condensed carbazole nuclei is found in a variety of indole alkaloids,we consider that the synthetic strategy described herein has possibility to be applied to other natural carbazole targets,further studies are ongoing and will be reported in due course.
AcknowledgmentsThe work was financially supported by NSFC (No. 21102026), Qiankehe [2011] 3002 and West Light Foundation of the Chinese Academy of Sciences.
Appendix A. Supplementary dataSupplementary data associated with this article can be found,in the online version,at http://dx.doi.org/10.1016/j.cclet.2014.11.026
| [1] | A.W. Schmidt, K.R. Reddy, H.J. Knölker, Occurrence, biogenesis, and synthesis of biologically active carbazole alkaloids, Chem. Rev. 112 (2012) 3193-3328. |
| [2] | C. Ito, H. Furukawa, New carbazole alkaloids from Murraya euchrestifolia Hayata, Chem. Pharm. Bull. 38 (1990) 1548-1550. |
| [3] | T.S. Wu, S.C. Huang, P.L. Wu, Pyrano-and furocarbazole alkaloids from the root bark of Clausena excavate, Heterocycles 45 (1997) 969-973. |
| [4] | For review see: W. Fröhner, M.P. Krahl, K.R. Reddy, et al., Transition metal complexes in organic synthesis, part 73, Synthetic routes to naturally occurring furocarbazoles, Heterocycles 63 (2004) 2393-2407. |
| [5] | Y. Xia, Z.X. Liu, Q. Xiao, et al., Rhodium(II)-catalyzed cyclization of bis(N-tosylhydrazone) s: an efficient approach towards polycyclic aromatic compounds, Angew. Chem. Int. Ed. 51 (2012) 5714-5717. |
| [6] | S.C. Pelly, C.J. Parkinson, W.A.L. van Otterlo, et al., Metathesis reactions for the synthesis of ring-fused carbazoles, J. Org. Chem. 70 (2005) 10474-10481. |
| [7] | S. Ohta, M. Okamoto, Synthesis of carbonyl compouds via biogenetic-type transamination reaction, Synthesis 9 (1982) 756-758. |
| [8] | A. Papanikos, J. Rademann, M. Meldal, a-Ketocarbonyl peptides: a general approach to reactive resin-bound intermediates in the synthesis of peptide isosteres for protease inhibitor screening on solid support, J. Am. Chem. Soc. 123 (2001) 2176-2181. |
| [9] | E.J. Corey, D.Y. Gin, R.S. Kania, Enantioselective total synthesis of ecteinascidin 743, J. Am. Chem. Soc. 118 (1996) 9202-9203. |
| [10] | S. Liu, Y.M. Cui, F.J. Nan, Total synthesis of the originally proposed and revised structures of scleritodermin A, Org. Lett. 10 (2008) 3765-3768. |
| [11] | C.J. Pearce, T.W. Doyle, S. Forenza, et al., The biosynthetic origins of rebeccamycin, J. Nat. Prod. 51 (1988) 937-940. |
| [12] | T. Nishizawa, S. Grüschow, D.H.E. Jayamaha, et al., Enzymatic assembly of the bis-Indole core of rebeccamycin, J. Am. Chem. Soc. 128 (2006) 724-725. |
| [13] | D.M. Tapiolas, B.F. Bowden, E. Abou-Mansour, et al., Eusynstyelamides A, B, and C, nNOS inhibitors, from the ascidian Eusynstyela latericius, J. Nat. Prod. 72 (2009) 1115-1120. |
| [14] | P. Grieco, Y.S. Hon, A. Perez-Medrano, Convergent, enantiospecific total synthesis of the novel cyclodepsipeptide (+)-jasplakinolide (jaspamide), J. Am. Chem. Soc. 110 (1988) 1630-1631. |
| [15] | N. Wahlström, I. Romero, J. Bergman, Synthesis of metabolites of the Ah receptor ligand 6-formylindolo[3, 2-b]carbazole, Eur. J. Org. Chem. 12 (2004) 2593-2602." |