




b State Key Laboratory of Bioorganic and Natural Products Chemistry, Shanghai Institute of Organic Chemistry, Chinese Academy of Sciences, Shanghai 200032, China
Acridone alkaloids comprise a large family of biologically active natural products [1],some of which display significant anticancer activity [2]. Chlorospermines A and B (1 and 2,Fig. 1) are two polycyclic acridone alkaloids recently isolated by Litaudon and co-workers from the stem bark of Glycosmis chlorosperma[3]. The latter possesses remarkable inhibitory property against dual-specificity tyrosine phosphorylation-regulated kinase 1A (DYRK1A),which is closely related to neuronal development and adult brain physiology. Although a number of acridone syntheses have been documented in literature [4, 5, 6, 7, 8, 9, 10, 11],they usually relied on Friedel-Crafts reactions which require strongly acidic conditions as well as electron-rich arene substrates. In order to build up a focused library of chlorospermine analogs for further biological evaluations,an efficient and modular approach to the tetracyclic core of chlorospermines is highly desired. This approach needs to be applicable to a wide range of substrates; thus,constructing the acridone core under neutral conditions would be optimal. The synthesis of multi-substituted arenes remains a remarkable challenge for modern organic chemistry [12, 13, 14, 15, 16, 17, 18, 19, 20, 21, 22, 23, 24]. Electrocyclization has proven to be a powerful tool for assembling functionalized ring systems,since Nicolaouet al.disclosed the elegant synthesis of endiandric acids [25, 26, 27, 28, 29, 30, 31, 32, 33, 34, 35, 36, 37, 38, 39, 40, 41, 42, 43]. Notably,multi-substituted arenes can be constructed in a highly efficient and convergent fashion,when 6pelectrocyclization is strategically combined with oxidative aromatization [44, 45, 46, 47, 48, 49, 50, 51, 52, 53, 54]. Recently,we have successfully applied such strategies to the syntheses of a series of natural products containing aromatic moieties,such as daphenylline [55],tubingensin A [56],xiamycin A,oridamycins A and B [57],rubriflordilactone A [58] and clostrubin [59]. Herein,we report a concise route toward the tetracyclic core of chlorospermines A and B using 6pelectrocyclization/aromatization as a key step.

|
Download:
|
| Fig. 1. The structures of chlorospermines A and B. | |
{kind=link}
All reactions were carried out under an argon atmosphere with dry solvents under anhydrous conditions,unless otherwise noted. Tetrahydrofuran (THF) was distilled immediately before use from sodium-benzophenone ketyl. Methylene chloride (CH2Cl2) and triethylamine (Et3N) were distilled from calcium hydride and stored under an argon atmosphere. Reagents were purchased at the highest commercial quality and used without further purification,unless otherwise stated. Solvents for chromatography were used as supplied by Titan chemical. Reactions were monitored by thin layer chromatography (TLC) carried out on S-2 0.25 mm E. Merck silica gel plates (60F-254) using UV light as visualizing agent and aqueous ammonium cerium nitrate/ammonium molybdate or basic aqueous potassium permanganate as developing agent. E. Merck silica gel (60,particle size 0.040- 0.063 mm) was used for flash column chromatography. NMR spectra were recorded on Bruker AV-400 instrument and calibrated by using residual undeuterated chloroform (dH7.26) and CDCl3 (dC 77.16) as internal references. IR spectra were recorded on a Thermo Scientific Nicolet 380 FT-IR spectrometer. High-resolution mass spectra (HRMS) were recorded on a Bruker APEXIII 7.0 Tesla ESI-FT.
The preparation of compound 3: To a stirred solution of known compound5(2.00 g,11.4 mmol) in DMF (10 mL) was added NIS (2.57 g,12.6 mmol) at 0℃. The reaction mixture was stirred at 22℃ for 1.5 h before it was diluted with EtOAc (80 mL). The resultant mixture was sequentially washed with saturated aq. Na2S2O3(20 mL) and brine (20 mL),and the organic phase was dried over anhydrous Na2SO4 and filtered. The solvent was evaporated under vacuum,and the crude iodide was dissolved in CH2Cl2(10 mL). To this solution were sequentially added TsCl (2.61 g,13.7 mmol),Et3N (5.76 g,7.93 mL,57.0 mmol),and 4-DMAP (139 mg,1.14 mmol) at 0℃. The resultant mixture was allowed to warm to 22℃ and stirred at that temperature for 1 h before it was quenched with saturated aq. NaHCO3(30 mL). The mixture so obtained was extracted with EtOAc (3×60 mL),and the combined organic phases were washed with brine (2×20 mL) and dried over anhydrous Na2SO4. After filtration and removal of the solvent under vacuum,the residue was subjected to flash column chromatography for purification using EtOAc/petroleum ether (1:5!1:1) as eluent to givea-iodoquinolone 3 (4.83 g,93% for the two steps) as a yellow foam (Scheme 1).3: IR (film,cm-1 ):nmax 3063,2929,2843,1593,1560,1483,1263,1180,1037,812,870, 757,677,570; 1HNMR (400 MHz,CDCl3):δ9.09 (s,1H),7.93 (d,2H, J=8.1 Hz),7.57 (d,1H,J=8.6 Hz),7.45 (t,1H,J=8.2 Hz),7.40 (d, 2H,J=8.1 Hz),7.10 (d,1H,J=7.8 Hz),4.08 (s,3 H),2.50 (s,3H); 13C NMR (101 MHz,CDCl3):δ155.99,155.09,154.48,146.16,141.03, 133.49,129.87,128.65,128.12,126.01,114.33,108.70,87.33, 56.08,21.67; HRMS (m/z ): [M+H]+ calcd. for C17H15O4NIS+ 455.9761,found 455.9763.

|
Download:
|
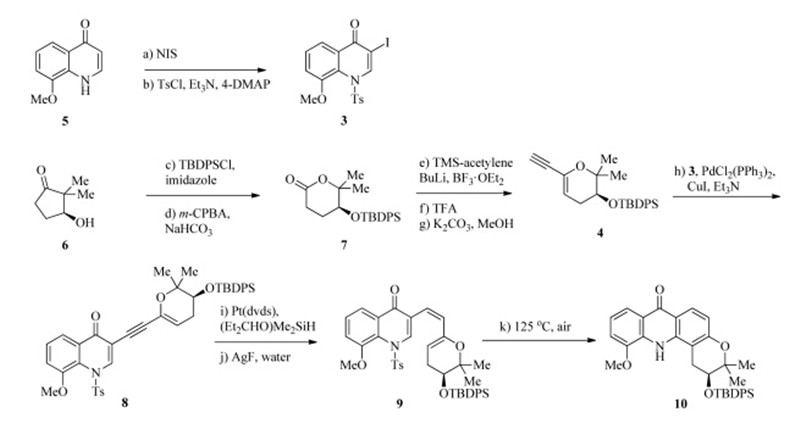
| Scheme 1.Synthesis of the tetracyclic core of chlorospermines A and B. Reagents and conditions: (a) NIS (1.1 equiv.),DMF,22℃,1.5 h; (b) TsCl (1.2 equiv.),Et3N (5.0 equiv.), 4-DMAP (10 mol%),CH2Cl2,22℃,1 h,93% for the two steps; (c) TBDPSCl (5.0 equiv.),imidazole (5.0 equiv.),DMF,22℃,1 h; (d)m-CPBA (1.3 equiv.),NaHCO3(1.3 equiv.), 22℃,5 h,87% for the two steps; (e) TMS-acetylene (4.5 equiv.),BuLi (3.0 equiv.),BF3·OEt2(3.0 equiv.),THF,-78℃,2 h; (f) TFA (0.5 equiv.),4 A˚ molecular sieves,CH2Cl2, 22℃,24 h; (g) K2CO3(2.0 equiv.),MeOH,22℃,2 h,53% for the three steps; (h) Pd(PPh3)2Cl2(5 mol%),CuI (5 mol%),Et3N (5.0 equiv.),3(1.0 equiv.),DMF,22℃,12 h,75%; (i) Pt(dvds) (5 mol%),(Et2CHO)Me2SiH (1.1 equiv.),CH2Cl2,22℃,10 h; (j) AgF (3.0 equiv.),THF/MeOH/water (500:250:1),22℃,3 h,41% for the two steps; (k) air,toluene, 125℃,3 h,56% | |
{kind=link}
The preparation of compound 7: To a stirred solution of known β-hydroxy ketone 6(2.00 g,15.6 mmol) in DMF (1.0 mL) were sequentially added imidazole (5.31 g,78.0 mmol) and TBDPSCl (21.4 g,20.3 mL,78.0 mmol) at 0℃. The reaction mixture was allowed to warmed to 22℃ and stirred at that temperature for 1 h before it was quenched with saturated aq. NaHCO3(30 mL). The resultant mixture was extracted with EtOAc (3×40 mL). The combined organic phases were washed with brine (50 mL),dried over anhydrous Na2SO4,and filtered. The solvent was evaporated under vacuum,and the residue was passed through a plug of silica with EtOAc/petroleum ether (1:20) to give the TBDPS ether as a colorless oil. This oil was dissolved in CH2Cl2 (15 mL). To this solution were sequentially added NaHCO3(1.70 g,20.3 mmol) and m-CPBA (4.12 g,20.3 mmol,85 wt%) at 0℃. The reaction mixture was allowed to warm to 22℃ and stir at that temperature for 5 h before it was quenched with saturated aq. Na2SO3(50 mL). The resultant mixture was extracted with CH2Cl2(3×60 mL),and the combined organic phases were washed with brine (80 mL) and dried over anhydrous Na2SO4. After filtration and removal of the solvent under vacuum,the residue was purified by flash column chromatography with EtOAc/petroleum ether (1:10!1:5) to give lactone7(5.19 g,87% for the two steps) as a colorless oil (Scheme 1).7: IR (film,cm-1 ): nmax3066,2959,2931,2857,1736,1474, 1429,1274,1114,1013,819,698; 1HNMR (400 MHz,CDCl3): d 7.70-7.65 (m,4H),7.48-7.44 (m,2H),7.42-7.38 (m,4H),3.78 (dd, 1H,J=6.0,5.0 Hz),2.61 (dd,1H,J=18.8,7.4 Hz),2.30 (dd,1H, J=18.8,7.0 Hz),1.81-1.76 (m,2H),1.42 (s,3H),1.31 (s,3H),1.08 (s, 9H); 13C NMR (101 MHz,CDCl3):δ170.29,135.79,135.71,133.47, 132.60,130.03,129.96,127.78,127.68,84.24,71.35,27.47,26.95, 25.98,24.31,23.98,19.37; HRMS (m/z ): [M+H]+ calcd. for C23H31O3Si+ 383.2037,found 383.2034.
The preparation of compound 4: To a stirred solution of (trimethylsilyl)acetylene (6.00 g,8.63 mL,61.0 mmol) in THF (120 mL) at-78℃ was added BuLi (17.0 mL,2.4 mol/L in hexane, 40.8 mmol). The resulting mixture was stirred at-78℃ for 15 min before BF3·OEt2 (5.79 g,5.03 mL,40.8 mmol) was added. The mixture so obtained was stirred at-78℃ for 30 min before a solution of 7 (5.19 g,13.6 mmol) in THF (18 mL) was added. The reaction mixture was stirred at -78℃ for 2 h before it was quenched with saturated aq. NH4Cl (100 mL). The resultant mixture was extracted with EtOAc (3×80 mL). The combined organic phases were washed with brine and dried over anhydrous Na2SO4. After filtration and removal of the solvent under vacuum, the crude alcohol was dissolved in CH2Cl2(24 mL). To this solution were sequentially added 4 Å molecular sieves (3.40 g) and TFA (775 mg,520mL,6.80 mmol) at 0℃. The reaction mixture was stirred at 22℃ for 24 h before it was quenched with saturated aq. NaHCO3(30 mL). The resultant mixture was extracted with CH2Cl2 (3×60 mL),and the combined organic phases were washed with brine (3×20 mL),dried over anhydrous Na2SO4,and filtered. The solvent was removed under vacuum,and the residue was dissolved in MeOH (30 mL). To this solution was added anhydrous K2CO3 (3.76 g,27.1 mmol) at 22℃. The resultant mixture was stirred at that temperature for 2 h before it was quenched with saturated aq. NH4Cl (30 mL). The mixture so obtained was extracted with EtOAc (3×50 mL). The combined organic phases were washed with brine (2×50 mL) and dried over anhydrous Na2SO4. After filtration and removal of the solvent under vacuum,the residue was subjected to flash column chromatography for purification using EtOAc/petroleum ether (1:100) as eluent to give pyran 4 (2.81 g,53% for the three steps) as a colorless oil (Scheme 1).4:IR (film,cm-1 ):nmax3286,3071,2934,2854,1640,1426,1313,1114, 837,742,703,617; 1HNMR (400 MHz,CDCl3):δ7.71-7.66 (m,4H), 7.47-7.43 (m,2H),7.41-7.37 (m,4H),5.02 (dd,1H,J=4.8,3.5 Hz), 3.68 (dd,1H,J=8.1,6.2 Hz),2.83 (s,1H),2.03-1.93 (m,2H),1.30 (s, 3H),1.29 (s,3H),1.07 (s,9H); 13C NMR (101 MHz,CDCl3):δ136.04, 136.03,134.25,134.04,133.25,130.02,129.86,127.85,127.64, 106.66,79.46,78.16,75.05,71.51,28.75,27.11,26.22,19.50, 19.01; HRMS (m/z ): [M+Na]+ calcd. for C25H30NaO2Si+ 413.1907, found 413.1902.
The preparation of compound 8: To a stirred solution of aiodoquinolone 3 (1.00 g,2.20 mmol) and pyran 4 (860 mg, 2.20 mmol) in DMF (3.0 mL) were sequentially added Pd(PPh3)2Cl2 (77.2 mg,0.110 mmol),CuI (20.9 mg,0.110 mmol),and Et3N (1.12 g,1.54 mL,11.0 mmol) at 22℃. The reaction mixture was stirred at that temperature for 12 h before it was quenched with saturated aq. NaHCO3 (10 mL). The mixture so obtained was extracted with EtOAc (3×30 mL). The combined organic phases were then washed with brine (50 mL),dried over anhydrous Na2SO4,and filtered. The solvent was removed under vacuum,and the residue was purified by flash column chromatography with EtOAc/petroleum ether (1:5!1:1) to give dieneyne 8(1.18 g,75%) as a pale yellow foam.8: IR (film,cm-1 ):nmax3071,2959,2929, 2852,2215,1748,1593,1480,1382,1176,1111,903,757,703, 668; 1HNMR (400 MHz,CDCl3):δ8.86 (s,1H),7.84 (d,2H, J=8.4 Hz),7.72-7.68 (m,4H),7.57 (dd,1H,J=1.1,8.6 Hz),7.48- 7.39 (m,7H),7.24 (d,2H,J=8.4 Hz),7.07 (dd,1H,J=7.8,0.8 Hz), 5.01 (dd,1H,J=5.0,3.4 Hz),4.07 (s,3H),3.70 (dd,1H,J=8.4, 6.2 Hz),2.34 (s,3H),2.13-1.99 (m,2H),1.34 (s,3H),1.33 (s,3H), 1.08 (s,9H); 13C NMR (101 MHz,CDCl3):δ155.28,153.32,151.86, 145.79,140.84,136.05,136.03,134.43,134.13,133.18,133.09, 130.08,130.01,129.93,128.84,128.21,127.89,127.69,124.34, 114.70,112.04,109.21,108.26,92.86,80.06,78.29,71.62,56.31, 29.18,27.11,26.43,21.87,19.50,18.79; HRMS (m/z ): [M+H]+ calcd. for C42H44O6NSSi+ 718.2653,found 718.2646.
The preparation of compound 9: To a stirred solution of dieneyne 8(22.0 mg,0.0306 mmol) and (Et2CHO)Me2SiH (4.9 mg, 0.034 mmol) in CH2Cl2 (0.20 mL) was added Karstedt’s catalyst (30mL,0.05 mol/L in polydimethylsiloxane,0.0015 mmol) at 22℃. The reaction mixture was allowed to stir at 22℃ for 10 h before it was passed through a short plug of celite with Et2O (10 mL). The filtrate was concentrated under vacuum,and the residue (the crude mixture of two alkenylsilane regioisomers) was dissolved in THF/MeOH (1.5 mL,2:1). To this solution were sequentially added water (ca. 2mL) and AgF (11.6 mg, 0.0918 mmol) at 0℃. The reaction mixture was allowed to warm to 22℃ and stir at that temperature for 3 h in the dark before it was passed through a short plug of celite with EtOAc (15 mL). The filtrate was washed with saturated aq. NH4Cl (2×3 mL) and dried over anhydrous Na2SO4. After removal of the solvent under vacuum,the residue was subjected to flash column chromatography for purification using Et3N/EtOAc/petroleum ether (1:20:80 then 3:100:200) as eluent to give cis-triene 9(9.0 mg,41% for the two steps) as a pale yellow oil. This compound is unstable and needs to be used for the next step immediately.9: IR (film,cm-1 ): nmax3069,2929,2857,2220,1596,1483,1382,1177,1111,1054, 909,703,549; 1HNMR (400 MHz,benzene-d6):δ9.01 (s,1H),7.98 (dd,1H,J=8.5,0.9 Hz),7.79-7.65 (m,7H),7.21-7.19 (m,4H),7.15- 7.12 (m,2H),6.55-6.53 (m,3H),5.96 (d,1H,J=12.2 Hz),5.31 (d, 1H,J=12.2 Hz),4.25 (dd,1H,J=5.2,3.1 Hz),3.55 (dd,1H,J=8.7, 5.8 Hz),3.51 (s,3H),1.98 (ddd,1H,J=17.8,8.7,3.1 Hz),1.88-1.77 (m,1H),1.75 (s,3H),1.21 (s,3H),1.07 (s,3H),0.90 (s,9H); the 13C NMR experiment was not performed because of the instability of the compound; HRMS (m/z ): [M+H]+ calcd. for C42H46O6NSSi+ 720.2810,found 720.2800.
The preparation of compound10: A solution of cis-triene 9 (9.0 mg,0.0125 mmol) in toluene (9.0 mL) was heated to 125℃ and stirred at that temperature for 3 h under an air atmosphere before it was cooled to 22℃. The volatile was removed under vacuum,and the residue was purified by flash column chromatography with EtOAc/petroleum ether (1:5!1:2) to give tetracyclic acridone10(3.9 mg,56%) as a white foam. 10: IR (film, cm-1 ):nmax3426,2926,2854,1620,1530,1441,1257,1111,1057, 816,763,700,611; 1HNMR (400 MHz,CDCl3): d8.70 (d,1H, J=8.9 Hz),8.52 (d,1H,J=7.6 Hz),7.76-7.73 (m,2H),7.65-7.63 (m, 2H),7.43 (s,1H),7.20-7.18 (m,3H),7.13-7.11 (m,3H),6.91 (t,1H, J=8.0 Hz),6.80 (d,1H,J=8.9 Hz),6.48 (dd,1H,J=7.8,1.0 Hz), 3.96 (dd,1H,J=7.2,5.6 Hz),3.32 (s,3H),2.33 (dd,1H,J=15.4, 7.2 Hz),2.24 (dd,1H,J=15.4,5.6 Hz),1.36 (s,3H),1.23 (s,3H), 1.12(s,9H); 13C NMR (101 MHz,CDCl3):δ176.85,157.01,146.94, 139.52,136.27,136.20,134.63,133.40,131.48,130.40,130.03, 127.94,127.40,122.97,120.73,119.34,116.63,113.26,111.11, 103.42,77.74,71.41,55.38,27.16,26.51,25.84,20.71,19.57; HRMS (m/z ): [M+H]+ calcd. for C35H38O4NSi+ 564.2565,found 564.2555. 3. Results and discussion
The synthesis commenced with the preparation of the two segments,a-iodoquinolone 3 and alkynylpyran 4. The former arose from known compound 5[60]. Iodination with NIS afforded the corresponding 2-iodoenone,which further underwentN-protection (TsCl,Et3N,4-DAMP) to furnish 3 in 93% overall yield. It should be noted that the bromo counterpart [61] of 3 was much less reactive for the next Sonogashira coupling reaction and thus abandoned. The synthesis of the pyran segment took advantage of a Baeyer-Villiger oxidation. Silylation of β-hydroxy ketone 6 followed by treatment with m-CPBA under buffered conditions provided lactone7in 87% yield (two steps). This compound was exposed to lithiated TMS-acetelene in the presence of BF3·OEt2 to give an unstable lactol intermediate,which was dehydrated with TFA to generate a conjugate eneyne [62]. Desilylation with K2CO3/MeOH delivered 4 (53% yield for the three steps).
With the two segments in hand,we investigated the construction of the tetra-substituted arene. Compounds 3 and 4 were forged together through Sonogashira coupling (Pd(PPh3)2Cl2,CuI, Et3N) to afford dieneyne 8 in 75% yield. A number of conditions were examined for the transformation of 8 into the corresponding cis-triene9. Unfortunately,the partial hydrogenation with Lindlar catalyst lacked reproducibility,and Zn/Cu couple reduction [63] suffered low reactivity. The triene product 9 was found to be unstable and thus needed to be prepared under mild conditions and purified rapidly. To our delight,8 smoothly underwent Ptcatalyzed hydrosilylation [Pt(dvds),(Et2CHO)Me2SiH] to give a regioisomeric mixture of alkenylsilanes [64, 65, 66],which were then subjected to desilylation conditions [67, 68, 69, 70] (AgF,water) to form 9 in 41% overall yield. Finally,6pelectrocyclization/ aromatization occurred smoothly under thermal conditions (125℃) in the presence of air,to furnish tetracyclic acridone 10 in 56% yield. This compound may serve as a common intermediate for the synthesis of a series of chlorospermine analogs. 4. Conclusion
We have developed a convergent strategy to construct the tetracyclic core of chlorospermines A and B. The readily available building blocks are assembled together by Sonogashira coupling. Acis-triene intermediate was then prepared through a sequence of hydrosilylation/desilylation. A one pot 6p-electrocyclization/ aromatization process formed the tetra-substituted arene. Further studies toward the total synthesis of chlorospermines A and B and analogs thereof are currently underway.
AcknowledgmentFinancial support was provided by Ministry of Science & Technology (No. 2013CB836900),National Natural Science Foundation of China (Nos. 21290180,21172235 and 21222202),and China Postdoctoral Science Foundation (No. 2014M561537,M.Y.).
| [1] | J.P. Michael, Quinoline, quinazoline, and acridone alkaloids, Nat. Prod. Rep. 25 (2008) 166-187. |
| [2] | P. Belmont, J. Bosson, T. Godet, M. Tiano, Acridine and acridone derivatives, anticancer properties and synthetic methods: where are we now? Anticancer Agents Med. Chem. 7 (2007) 139-169. |
| [3] | M.A. Beniddir, E.L. Borgne, B.I. Iorga, et al., Acridone alkaloids from Glycosmis chlorosperma as DYRK1A inhibitors, J. Nat. Prod. 77 (2014) 1117-1122. |
| [4] | P.L. Macdonald, A.V. Robertson, The structure of acronycine, Aust. J. Chem. 19 (1966) 275-281. |
| [5] | J. Hlubucek, E. Ritchie, W.C. Taylor, A synthesis of acronycine, Aust. J. Chem. 23 (1970) 1819-1881. |
| [6] | D.G. Loughhead, Synthesis of des-N-methylacronycine and acronycine, J. Org. Chem. 55 (1990) 2245-2246. |
| [7] | A. Elomri, S. Michel, F. Tillequin, M. Koch, A novel synthesisi of 6-demethoxyacronycine, Heterocycles 34 (1992) 799-806. |
| [8] | R.C. Anand, N. Selvapalam, A practical regiospecific approach towards acronycine and related alkaloids, Chem. Commun. (1996) 199-200. |
| [9] | I.K. Kostakis, P. Magiatis, N. Pouli, et al., Design, synthesis, and antiproliferative activity of some new pyrazole-fused amino derivatives of the pyranoxanthenone, pyranothioxanthenone, and pyranoacidone ring systems: a new class of cytotoxic agents, J. Med. Chem. 45 (2002) 2599-2609. |
| [10] | S.L. MacNeil, B.J. Wilson, V. Snieckus, Anionic N-fries rearrangement of N-carbamoyl diarylamines to anthranilamides. Methodology and application to acridone and pyranoacridone alkaloids, Org. Lett. 8 (2006) 1133-1136. |
| [11] | G.S. Hari, Y.R. Lee, X. Wang, W.S. Lyoo, S.H. Kim, New synthetic routes to acronycine, noracronycine, and their analogues, Bull. Korean Chem. Soc. 31 (2010) 2406-2409. |
| [12] | K.P.C. Vollhardt, Cobalt-mediated [2 + 2+ 2]-cycloadditions: a maturing synthetic strategy, Angew. Chem. Int. Ed. 23 (1984) 539-644. |
| [13] | V. Gandon, C. Aubert, M. Malacria, Recent progress in cobalt-mediated [2 + 2 + 2] cycloaddition reactions, Chem. Commun. (2006) 2209-2217. |
| [14] | G. Domínguez, J. Pérez-Castells, Recent advances in [2 + 2 + 2] cycloaddition reactions, Chem. Soc. Rev. 40 (2011) 3430-3444. |
| [15] | D.L. Boger, Diels-Alder reactions of heterocyclic azadienes: scope and applications, Chem. Rev. 86 (1986) 781-793. |
| [16] | A.B. Smith, N.J. Liverton, N.J. Hrib, H. Sivaramakrishnan, K. Winzenberg, Total synthesis of (+)-jatropholones A and B. Exploitation of the high-pressure technique, J. Am. Chem. Soc. 108 (1986) 3040-3048. |
| [17] | P.S. Baran, N.Z. Burns, Total synthesis of (±)-haouamine A, J. Am. Chem. Soc. 128 (2006) 3908-3909. |
| [18] | Y. Liu, K. Lu, M. Dai, et al., An efficient one-pot asymmetric synthesis of biaryl compounds via Diels-Alder/retro-Diels-Alder cascade reactions, Org. Lett. 9 (2007) 805-808. |
| [19] | B.M. O'Keefe, D.M. Mans, D.E. Kaelin Jr., S.F. Martin, Total synthesis of isokidamycin, J. Am. Chem. Soc. 132 (2010) 15528-15530. |
| [20] | R.L. Greenaway, C.D. Campbell, O.T. Holton, C.A. Russell, E.A. Anderson, Palladium-catalyzed cascade cyclization of ynamides to azabicycles, Chem. Eur. J. 17 (2011) 14366-14370. |
| [21] | S.S. Goh, H. Baars, B. Gockel, E.A. Anderson, Metal-catalyzed syntheses of abridged CDE rings of rubriflordilactones A and B, Org. Lett. 14 (2012) 6278-6281. |
| [22] | C.D. Campbell, R.L. Greenaway, O.T. Holton, H.A. Chapman, E.A. Anderson, Palladium-catalyzed cyclization of bromoenynamides to tricyclic azacycles: synthesis of trikentrin-like frameworks, Chem. Commun. 50 (2014) 5187-5189. |
| [23] | Z. Lu, M. Yang, P. Chen, X. Xiong, A. Li, Total synthesis of hapalindole-type natural products, Angew. Chem. Int. Ed. 53 (2014) 13840-13844. |
| [24] | X. Xiong, D. Zhang, J. Li, et al., Synthesis of indole terpenoid mimics via a functionality-tolerated Eu(fod)3-catalyzed conjugate addition, Chem. Asian J. (2014), http://dx.doi.org/10.1002/asia.201403312. |
| [25] | K.C. Nicolaou, N.A. Petasis, R.E. Zipkin, J. Uenishi, The endiandric acid cascade. Electrocyclizations in organic synthesis. 1. Stepwise, stereocontrolled total synthesis of endiandric acids A and B, J. Am. Chem. Soc. 104 (1982) 5555-5557. |
| [26] | K.C. Nicolaou, N.A. Petasis, J. Uenishi, R.E. Zipkin, The endiandric acid cascade. Electrocyclizations in organic synthesis. 2. Stepwise, stereocontrolled total synthesis of endiandric acids C-G, J. Am. Chem. Soc. 104 (1982) 5557-5558. |
| [27] | K.C. Nicolaou, R.E. Zipkin, N.A. Petasis, The endiandric acid cascade. Electrocyclizations in organic synthesis. 3. ""Biomimetic"" approach to endiandric acids A-G. Synthesis of precursors, J. Am. Chem. Soc. 104 (1982) 5558-5560. |
| [28] | K.C. Nicolaou, N.A. Petasis, R.E. Zipkin, The endiandric acid cascade. Electrocyclizations in organic synthesis. 4. Biomimetic approach to endiandric acids A-G. Total synthesis and thermal studies, J. Am. Chem. Soc. 104 (1982) 5560-5562. |
| [29] | C.M. Beaudry, J.P. Malerich, D. Trauner, Biosynthetic and biomimetic electrocyclizations, Chem. Rev. 105 (2005) 4757-4778. |
| [30] | K.A. Parker, Y.-H. Lim, The total synthesis of (-)-SNF4435 C and (+)-SNF4435 D, J. Am. Chem. Soc. 126 (2004) 15968-15969. |
| [31] | C.M. Beaudry, D. Trauner, Total synthesis of (-)-SNF4435 C and (+)-SNF4435 D, Org. Lett. 7 (2005) 4475-4477. |
| [32] | A.K. Miller, D. Trauner, Mining the tetraene manifold: total synthesis of complex pyrones from Placobranchus ocellatus, Angew. Chem. Int. Ed. 44 (2005) 4602-4606. |
| [33] | P. Sharma, D.J. Ritson, J. Burnley, J.E. Moses, A synthetic approach to kingianin A based on biosynthetic speculation, Chem. Commun. 47 (2011) 10605-10607. |
| [34] | G.A. Barcan, A. Patel, K.N. Houk, O. Kwon, A torquoselective 6p electrocyclization approach to reserpine alkaloids, Org. Lett. 14 (2012) 5388-5391. |
| [35] | H.N. Lim, K.A. Parker, Total synthesis of kingianin A, Org. Lett. 15 (2013) 398-401. |
| [36] | S.L. Drew, A.L. Lawrence, M.S. Sherburn, Total synthesis of kingianins A, D, and F, Angew. Chem. Int. Ed. 52 (2013) 4221-4224. |
| [37] | C. Li, E. Lobkovsky, J.A. Porco, Total synthesis of (±)-torreyanic acid, J. Am. Chem. Soc. 122 (2000) 10484-10485. |
| [38] | H.M. Sklenicka, R.P. Hsung, M.J. McLaughlin, L.-l. Wei, A.I. Gerasyuto, W.B. Brennessel, Stereoselective formal [3 + 3] cycloaddition approach to cis-1-azadecalins and synthesis of (-)-4a, 8a-diepi-pumiliotoxin C. Evidence for the first highly stereoselective 6π-electron electrocyclic ring closures of 1-azatrienes, J. Am. Chem. Soc. 124 (2002) 10435-10442. |
| [39] | C. Li, R.P. Johnson, J.A. Porco, Total synthesis of the quinone epoxide dimer (+)-torreyanic acid: application of a biomimetic oxidation/electro-cyclization/Diels-Alder dimerization cascade, J. Am. Chem. Soc. 125 (2003) 5095-5106. |
| [40] | M. Volgraf, J.P. Lumb, H.C. Brastianos, et al., Biomimetic synthesis of the IDO inhibitors exiguamine A and B, Nat. Chem. Biol. 4 (2008) 535-537. |
| [41] | M. Chaumontet, R. Piccardi, O. Baudoin, Synthesis of 3,4-dihydroisoquinolines by a C(sp3)-H activation/electrocyclization strategy: total synthesis of coralydine, Angew. Chem. Int. Ed. 48 (2009) 179-182. |
| [42] | D.L. Sloman, J.W. Bacon, J.A. Porco, Total synthesis and absolute stereochemical assignment of kibdelone C, J. Am. Chem. Soc. 133 (2011) 9952-9955. |
| [43] | R. Hayashi, Z.X. Ma, R.P. Hsung, A tandem 1,3-H-shift-6p-electrocyclizationcyclic 2-amido-diene intramolecular Diels-Alder cycloaddition approach to BCD-ring of atropurpuran, Org. Lett. 14 (2012) 252-255. |
| [44] | E.A. Anderson, E.J. Alexanian, E.J. Sorensen, Synthesis of the furanosteroidal antibiotic viridin, Angew. Chem. Int. Ed. 43 (2004) 1998-2001. |
| [45] | K. Ohmori, K. Mori, Y. Ishikawa, H. Tsuruta, S. Kuwahara, N. Harada, K. Suzuki, Concise total synthesis and structure assignment of TAN-1085, Angew. Chem. Int. Ed. 43 (2004) 3167-3171. |
| [46] | D.C. Harrowven, D.D. Pascoe, D. Demurtas, H.O. Bourne, Total synthesis of (-)-colombiasin A and (-)-elisapterosin B, Angew. Chem. Int. Ed. 44 (2005) 1221-1222. |
| [47] | A. Fürstner, M.M. Domostoj, B. Scheiper, Total synthesis of dictyodendrin B, J. Am. Chem. Soc. 127 (2005) 11620-11621. |
| [48] | T.J. Greshock, R.L. Funk, Synthesis of indoles via 6p-electrocyclic ring closures of trienecarbamates, J. Am. Chem. Soc. 128 (2006) 4946-4947. |
| [49] | T.J. Greshock, R.L. Funk, An approach to the total synthesis of welwistatin, Org. Lett. 8 (2006) 2643-2645. |
| [50] | R.J. Huntley, R.L. Funk, Total syntheses of (±)-cis-trikentrin A and (±)-cis-trikentrin B via electrocyclic ring closures of 2,3-divinylpyrrolines, Org. Lett. 8 (2006) 3403-3406. |
| [51] | R.J. Huntley, R.L. Funk, A strategy for the total synthesis of dragmacidin E. Construction of the core ring system, Org. Lett. 8 (2006) 4775-4778. |
| [52] | S.T. Staben, J.J. Kennedy-Smith, D. Huang, et al., Gold(I)-catalyzed cyclizations of silyl enol ethers: application to the synthesis of (+)-lycopladine A, Angew. Chem. Int. Ed. 45 (2006) 5991-5994. |
| [53] | T. Suzuki, T. Hamura, K. Suzuki, Ring selectivity: successive ring expansion of two benzocyclobutenes for divergent access to angular and linear benzanthraquinones, Angew. Chem. Int. Ed. 47 (2008) 2248-2252. |
| [54] | D.L. Sloman, B. Mitasev, S.S. Scully, J.A. Beutler, J.A. Porco, Synthesis and biological evaluation of ABCD ring fragments of the kibdelones, Angew. Chem. Int. Ed. 50 (2011) 2511-2515. |
| [55] | Z. Lu, Y. Li, J. Deng, A. Li, Total synthesis of the daphniphyllum alkaloid daphenylline, Nat. Chem. 5 (2013) 679-684. |
| [56] | M. Bian, Z. Wang, X. Xiong, et al., Total syntheses of anominine and tubingensin A, J. Am. Chem. Soc. 134 (2012) 8078-8081. |
| [57] | Z. Meng, H. Yu, L. Li, et al., Total synthesis and antiviral activity of indolosesquiterpenoids from the xiamycin and oridamycin families, Nat. Commun. 6 (2015) 6096, http://dx.doi.org/10.1038/ncomms7096. |
| [58] | J. Li, P. Yang, M. Yao, J. Deng, A. Li, Total synthesis of rubriflordilactone A, J. Am. Chem. Soc. 136 (2014) 16477-16480. |
| [59] | M. Yang, J. Li, A. Li, Total synthesis of clostrubin, a potent antibiotic from Clostridium, Nat Commun. 6 (2015), http://dx.doi.org/10.1038/ncomms7445. |
| [60] | L. Hill, S.H. Imam, H. McNab, W.J. O'Neill, Regioselective synthesis of quinolin-4-ones by pyrolysis of anilinomethylene derivatives of Meldrum's acid, Synlett (2009) 1847-1850. |
| [61] | H. Yu, C. Wan, J. Han, A. Li, A protocol for a-bromination of b-substituted enones, Acta Chim. Sin. 71 (2013) 1488-1491. |
| [62] | M. Adachi, H. Yamada, M. Isobe, T. Nishikawa, Total synthesis of polygalolide A, Org. Lett. 13 (2011) 6532-6535. |
| [63] | A. Francais, A. Leyva, G. Etxebarria-Jardi, S.V. Ley, Total synthesis of the antiapoptotic agents iso-and bongkrekic acids, Org. Lett. 12 (2010) 340-343. |
| [64] | J.L. Speier, J.A. Webster, G.H. Barnes, The addition of silicon hydrides to olefinic double bonds. Part II. The use of group VIII metal catalysts, J. Am. Chem. Soc. 79 (1957) 974-979. |
| [65] | D.A. Rooke, E.M. Ferreira, Platinum-catalyzed hydrosilylations of internal alkynes: harnessing substituent effects to achieve high regioselectivity, Angew. Chem. Int. Ed. 51 (2012) 3225-3230. |
| [66] | D.A. Rooke, Z.A. Menard, E.M. Ferreira, An analysis of the influences dictating regioselectivity in platinum-catalyzed hydrosilylations of internal alkynes, Tetrahedron 70 (2014) 4232-4244. |
| [67] | A. Fürstner, K. Radkowski, A chemo-and stereoselective reduction of cycloalkynes to (E)-cycloalkenes, Chem. Commun. (2002) 2182-2183. |
| [68] | F. Lacombe, K. Radkowski, G. Seidel, A. Fürstner, (E)-Cycloalkenes and (E,E)-cycloalkadienes by ring closing diyne-or enyne-yne metathesis/semi-reduction, Tetrahedron 60 (2004) 7315-7324. |
| [69] | A. Fürstner, M. Bonnekessel, J.T. Blank, et al., Total synthesis of myxovirescin A1, Chem. Eur. J. 13 (2007) 8762-8783. |
| [70] | Y. Wang, M. Jimenez, A.S. Hansen, et al., Control of olefin geometry in macrocyclic ring-closing metathesis using a removable silyl group, J. Am. Chem. Soc. 133 (2011) 9196-9199." |