




Natural products have played an important role in the discovery and development of clinically useful antitumor agents [1]. It is reported that 79.8% of the marketed antitumor agents are derived from natural products or their synthetic derivatives from the year 1981 to 2010 [2]. For example,vinblastine,vincristine, campthothecin derivatives (e.g.topotecan and irinotecan),etoposide and paclitaxel are widely used in current antitumor chemotherapy. Currently,natural products inspired novel antitumor drug discovery continues to be an active area of research interests [3]. Numerous antitumor natural products have been reported and several of them (e.g.flavopiridol and combretastin A4 phosphate) are under clinical evaluation.
Evodiamine is a quinazolinocarboline alkaloid isolated from the fruit ofEvodia rutaecarpaBentham. Evodiamine was reported to be a multi-targeting antitumor lead compound with cytotoxicity against various human cancer cell lines [4,5]. In our previous studies,topoisomerase I (Top1) was identified as one of molecular target of evodiamine by a structure-based virtual screening study [6]. Moreover,a number of evodiamine derivatives were designed and synthesized [7]. Several evodiamine derivatives,such as 3-fluoroevodiamine (2)and 10-hydroxylevodiamine (3),showed excellent antitumor activity against a variety of cancer cell-lines. Furthermore,in silico target identification in combination with biological assays confirmed that these highly active evodiamine derivatives acted by dual inhibition of topoisomerases I and II [7].
Chirality is a key feature of natural products,and stereochemistry is often important for a specific biological activity [8]. Evodiamine and its derivatives have an asymmetric center at the C13b position (Fig. 1). It is interesting to know the effects of the C13b chiral center on the antitumor activity. Herein,isomers of evodiamine derivatives 2 and3 were obtained by asymmetric synthesis and their antitumor activity was investigated. 2. Experimental
Nuclear magnetic resonance (NMR) spectra were generated on a Bruker AVANCE300 and AVANCE500 spectrometer (Bruker Company,Germany),using CDCl3 as the reference standard or DMSO-d6. Chemical shifts (δ values) and coupling constants (j values) are expressed in ppm and Hz,respectively. ESI mass spectra were gathered on an API-3000 LC-MS spectrometer. TLC analysis was carried out on silica gel plates GF254 (Qindao Haiyang Chemical,China). Silica gel column chromatography was performed with Silica gel 60 G (Qindao Haiyang Chemical,China). Commercial solvents were used without any pretreatment.

|
Download:
|
| Fig. 1. Chemical structures of evodiamine and its derivatives | |
Generally,the isomers of evodiamine derivatives2 and3were prepared according to the method described in Scheme 1. The anilines 4a-b were treated with NaNO2 to yield diazkonium compounds5a-b. Hydrolysis of ethyl 2-oxopiperidine-3-carboxylate (6) gave the compound 7. The key b-carboline intermediates 9a-b were synthesized by reacting5a-bwith7,followed by treating with HCOOH at reflux [9]. In the presence of POCl3,intermediates 9a-b were reacted with methyl 2-(methylamino)benzoate 10a-b in anhydrous THF to afford the dehydroevodiamine derivatives 11a-b . Finally,asymmetric catalytic hydrogenation of 11a-b by RuCl[(S,S)-Tsdpen](p-cymene) or RuCl[(R,R)-Tsdpen](p-cymene) gave (S)-2,(S)-12,(R)-2 and (R)-12 using Noyori’s procedure [10]. After removal of the protection group,compounds (S)-3 and (R)-3 were obtained. The absolute configuration of the isomers were assigned according to the previous reports [7,10].

|
Download:
|
| Scheme 1.Synthetic routes of the target compounds. Reagents and conditions: (a) NaNO2,hydrochloric acid,H2O,0-5℃,2 h,yield 90%; (b) KOH,H2O,r.t.,12 h,yield 93%; (c) r.t.,10 h,yield 85%; (d) HCOOH,reflux,0.5 h,yield 55%-59%; (e) POCl3,THF,reflux,3 days,yield 55%; (f) Cat. A,HCOOH:Et3N (5:2),0℃,8 h,yield 78%-87%; (g) Cat. B, HCOOH:Et3N (5:2),0℃,8 h,yield 84%; (h) 10% Pd/C,H2,DMF,r.t.,12 h,yield 83%-87%. | |
To a stirring solution of 9a[7] (0.3 g,1.6 mmol) in dry THF (20 mL),POCl3(0.22 mL,2.4 mmol) was added,and the mixture was stirred under nitrogen atmosphere at 60℃. After 40 min, methyl 5-fluoro-2-(methylamino)benzoate (0.45 g,2.4 mmol) was added and the reaction mixture was stirred under reflux for 3 days. The mixture was diluted with water (100 mL) and then extracted with EtOAc (3×100 mL). The combined organic layers were washed with saturated sodium chloride solution (3×100 mL), dried over anhydrous Na2SO4 and concentrated under reduced pressure. The residue was purified by column chromatography (gradient CH2Cl2:MeOH = 100:2-100:5) to give compound 11a as a yellow solid (0.29 g,yield 55%). 1H NMR (DMSO-d6,600 MHz): δ 3.32 (t,2H,J= 6.6 Hz),4.04 (s,3H),4.64 (t,2H,J= 6.6 Hz),7.25 t,1H,J= 7.5 Hz),7.51 (t,1H,J= 7.5 Hz),7.70 (d,1H,J= 8.2 Hz),7.86 (d,1H,J= 8.2 Hz),8.01-8.05 (m,1H),8.09 (dd,1H,J= 7.8 Hz, 3.1 Hz),8.28 (dd,1H,J= 9.0 Hz,3.9 Hz),12.78 (s,1H). MS (ESI, positive)m/z calcd. for C19H15FN3O+ (M): 320.34; found: 320.12.
The synthetic method for compound11bwas similar to that of compound11a. 2.2. Synthesis of (S)-3-fluoroevodiamine ((S)-2)
To a stirred solution of 11a(49 mg,0.14 mmol) and RuCl[(S,S)-Tsdpen](p-cymene) (4 mg,6 mmol%) in DMF (2.5 mL) was added the HCO2H-triethylamine mixture (5:2,18mL). Then,the reaction mixture was stirred at 0℃ for 8 h,diluted with water (15 mL),and extracted with EtOAc (3×15 mL). The organic layer was dried over Na2SO4and the solvent was evaporated in vacuo. The residue was purified by column chromatography (hexane:EtOAc = 4:1) to give compound (S)-2 as a white solid (35 mg,yield 78%). 1H NMR (DMSO-d6,500 MHz): δ2.65 (s,3H),2.83-2.84 (m,2H),3.16-3.21 (m,1H),4.59-4.62 (m,1H),6.07 (s,1H),7.00 (t,1H,J= 7.7 Hz),7.10 (t,1H,J= 8.3 Hz),7.18 (dd,1H,J= 8.8 Hz,4.4 Hz),7.73-7.38 (m, 2H),7.48 (d,1H,J= 7.7 Hz),7.52 (dd,1H,J= 8.8 Hz,3.3 Hz),11.15 (s,1H). 13CNMR (DMSO-d6,75 MHz): δ19.6,36.6,69.2,111.6, 113.2,113.5,118.3,118.9,120.4,120.7,122.0,125.8,129.6,136.7, 146.0,155.9,158.7,163.0. MS (ESI,positive) m/z calcd. for C19H17FN3O (M+H): 322.36; found: 322.59. HPLC (Daicel Chiralpak AD,0.46 cm I.D.×25 cm L×5mm,25℃,i-propanol/hexane = 30/ 70,flow rate = 0.8 mL/min,λ= 254 nm): tmajor= 8.72 min,tminor== 6.51 min,ee= 99%.a22D 453.8 (c1.45,MeOH).
The synthetic method for target compounds (R)-2,(S)-12,(R)-12 was similar to that of compound (S)-2.
(R)-3-Fluoroevodiamine ((R)-2): White solid: 38 mg (yield: 84%). HPLC (Daicel Chiralpak AD,0.46 cm I.D.×25 cm L×5mm,25℃,ipropanol/hexane = 30/70,flow rate = 0.8 mL/min,λ=254 nm): tmajor= 6.29 min,tminor== 8.34 min,ee= 92%. [a] 22D -500:7(c 1.45 mg/mL,MeOH).
(S)-10-Benzyloxyevodiamine (,(S)-12): Yellow solid: 40 mg (yield: 87%). 1H NMR (CDCl3,500 MHz) d: 2.51 (s,3H),2.90- 2.93 (m,2H),3.25-3.30 (m,1H),4.84-4.89 (m,1H),5.12 (s,2H), 5.89 (s,1H),6.98 (dd,1H,J= 9.0 Hz,2.4 Hz),7.10 (s,1H),7.14 (d, 1H,J= 8.4 Hz),7.20 (t,1H,J= 7.5 Hz),7.30-7.51 (m,7H),8.12 (dd, 1H,J= 7.2 Hz,1.5 Hz),8.19 (s,1H). MS (ESI,negative)m/z calcd. for C26H22N3O2(M-H): 408.47; found: 408.59. 2.3. Synthesis of (S)-10-hydroxyevodiamine ((S)-3
A solution of (S)-12(30 mg,0.07 mmol) and 10% Pd/C (3 mg) in EtOAc (10 mL) was stirred under hydrogen atmosphere at room temperature for 12 h. The mixture was filtered,and then the solvent was evaporated under reduced pressure. The residue was purified by column chromatography (Hexane: EtOAc = 3:1) to give compound (S)-3 as a yellow solid (20 mg,yield 83%). 1H NMR (DMSO-d6,500 MHz):d2.65-2.68 (m,1H),2.80-2.89 (m,1H),2.89 (s,3H),3.15-3.19 (m,1H),4.58-4.62 (m,1H),6.08 (s,1H),6.61- 6.62 (d,1H,J= 8.5 Hz),6.75 (s,1H),6.94 (t,1H,J= 7.5 Hz),7.02 (d, 1H,J= 8.2 Hz),7.14 (d,1H,J= 8.5 Hz),7.46 (t,1H,J= 7.5 Hz),7.78 (d,1H,J= 7.8 Hz),8.68 (s,1H),10.70 (s,1H). 13CNMR (DMSO-d6, 75 MHz):d20.0,31.1,36.8,70.4,102.6,111.5,112.5,112.5,117.5, 119.5,120.5,127.1,128.4,131.4,131.6,133.9,149.1,151.1, 164.7. MS (ESI,positive)m/z calcd. for C19H18N3O2(M+H): 320.37; found: 320.14. HPLC (Chiralpak AD,0.46 cm I.D.×25 cm L×5mm, 25℃,i-propanol/hexane = 30/70,flow rate 0.6 mL/min, λ=254 nm):tmajor= 4.399 min,ee>99%.[a] 22D 316.1 (c2.5,MeOH) The synthetic method for target compounds (R)-3was similar to that of compound (S)-3.
(R)-10-Hydroxyevodiamine ((R)-3): Yellow solid: 21 mg (yield: 87%). HPLC (Chiralpak AD,0.46 cm I.D.×25 cm L×5mm,25℃, i-propanol/hexane = 30/70,flow rate 0.6 mL/min,λ=254 nm): tmajor= 6.605 min,tminor== 4.459 min,ee= 91%.[a] 22D -283.0 (c2.5, MeOH). 2.4. Top1-mediated supercoiled pBR322 relaxation assay
Relaxation assays were carried out as described [7]. The 20mL final reaction buffer contained 35 mmol/L Tris-HCl (pH 8.0), 72 mmol/L KCl,5 mmol/L MgCl2,5 mmol/L dithiothreitol,5 mmol/ L spermidine,0.1% bovine serum albumin (BSA),0.25mg pBR322 plasmid DNA and 1 unit of Top1 (TaKaRa Biotechnology Co.,Ltd., Dalian),1mL of different compounds solution or the Top1 inhibitor (CPT) as positive control. The reaction mixture was incubated at 37℃ for 15 min and terminated by the addition of 2mLof10× loading buffer (0.9% sodium dodecyl sulfate (SDS),0.05% bromophenol blue,and 50% glycerol). Then the DNA samples were subjected to electrophoresis in 0.8% agarose gel in 1×TAE (Tris- acetate-EDTA) at 8 V/cm for 1 h. Gels were stained with ethidium bromide (0.5mg/mL) for 60 min. The DNA band was visualized over UV light and photographed with Gel Doc Ez imager (Bio-Rad Laboratories Ltd.). 2.5. Top2-mediated supercoiled pBR322 relaxation assay
Relaxation assays were performed using Topoisomerase II Drug Screening Kit (TopoGEN,Inc.) [7]. The 20mL final reaction mixture contained 50 mmol/L Tris-HCl (pH 8.0),150 mmol/L NaCl, 10 mmol/L MgCl2,5 mmol/L dithiothreitol,30mg/mL bovine serum albumin (BSA),2 mmol/L ATP,0.25mg pBR322 plasmid DNA, 0.75 unit of Top2 (TopoGEN,Inc.),and 1mL of different compounds solution or the Top2 inhibitor (Eto) as positive control. The reaction mixture was incubated at 37℃ for 30 min and then terminated by the addition of 2mL10×gel loading buffer (0.25% bromophenol blue,50% glycerol). The samples were analyzed on 1% agarose gel at 8 V/cm for 1 h with 1×TAE (Tris-acetate-EDTA) as the running buffer. Gels were stained with ethidium bromide (0.5mg/mL) for 60 min. The DNA band was visualized over UV light and photographed with Gel Doc Ez imager (Bio-Rad Laboratories Ltd.). 2.6. In vitro cytotoxicity assay
Cells were seeded in 96-well microtiter plates at a density of 5×103 /well in a humidified atmosphere with 5% CO2at 37℃ for 24 h. All compounds to be tested were dissolved in DMSO at a concentration of 10 mmol/L. Cells were treated in triplicate with gradient concentrations of compounds at 37℃ for 72 h,with a 5% CO2environment. Then,20mL of MTT (3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide) solution (5 mg/mL) was added to each well. After incubated for an additional 4 h,the formazan was dissolved in 100mL of DMSO. The absorbance (OD) was read on a WellscanMK-2 microplate reader (Labsystems) at 570 nm. The concentration causing 50% inhibition of cell growth (IC50) was determined by the Logit method [11,12]. All experiments were performed three times. 2.7. Molecular docking
The X-ray crystallographic structures of CPT-DNA-Top1 ternary complex (PDB code: 1T8I [13]) and ATPase domain of human Top2a(PDB code: 1ZXM [14]) were downloaded from the protein data bank and prepared for docking using protein prepare protocol in Discovery Studio 3.0 [15]. During this process,the ligand was removed from the binding pocket,and polar hydrogens were added. All the waters were deleted and each atom was rendered with Gasteiger charges. In silico docking was carried out using GOLD 5.1 [16] on a Linux PC as described before [6]. 3. Results and discussion 3.1. In vitro antitumor activity
The growth inhibitory activities toward human cancer cell-lines A549 (lung cancer),HCT116 (colon cancer),ZR-75-30 (human breast cancer),CNE (human thyroid carcinoma) and KYSE-150 (human esophageal squamous cell carcinoma) were determined using MTT assay. As shown in Table 1,all the four isomers showed good to excellent antitumor activity against the five tested cancer cell lines. Particularity,these isomers were highly active toward A549 and HCT116 cell lines with IC50values in the range of 0.004- 0.02mmol/L. Interestingly,it can be seen that the C13b chiral center played an important role for the antitumor potency. In general,the (S)-isomers showed better antitumor activity than the (R)-isomers. However,the effects were different for specific compound and cancer cell lines. For the A549 cell line,(S)-2and (S)-3was 3.5 fold and 2 fold more potent than (R)-2and (R)-3, respectively. In contrast,the difference of the activity was enlarged for the HCT116 cell line. (S)-2 and (S)-3was 11.4 fold and 4.9 fold more potent than (R)-2and (R)-3,respectively. However,reverse trend was observed for isomers 3for the ZR-75-30 cell line and isomers2for the CNE cell line. In both cases,the (R)-isomers was more active than the (S)-isomers. For the (S)-isomers,(S)-3was more active than (S)-2toward the A549,ZR-75-30 and CNE cell lines,whereas (S)-2showed better activity than (S)-3toward the KYSE-150 cell line.
| Table 1 In vitro antitumor activity of isomers of evodiamine derivatives (IC50,mmol/L) |

{kind=link}
{kind=link}
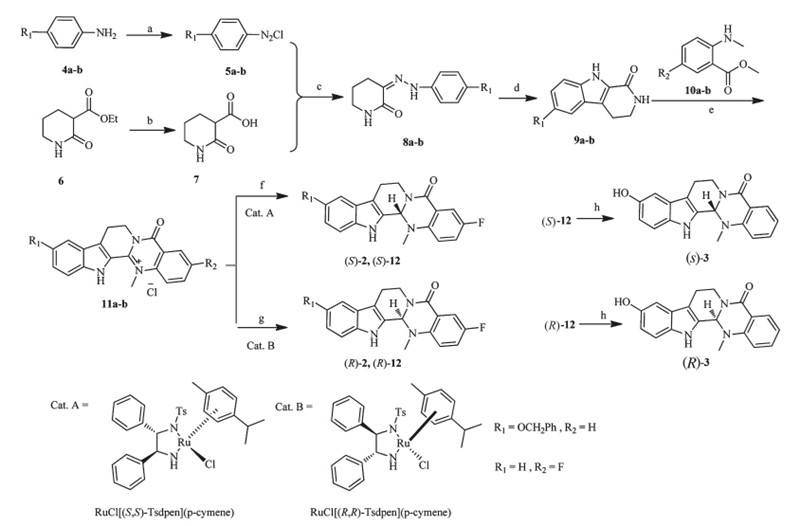
In the previous study [7,17],we found evodiamine and its derivatives were dual Top1/Top2 inhibitors. To investigate the difference of Top1 and Top2 inhibitory activity between these isomers,Top1- and Top2-mediated pBR322 DNA relaxation assays were performed. Camptothecin (CPT,a Top1 inhibitor) and etoposide (Eto,a Top2 inhibitor) were used as the positive controls and drugs were dosed at 50mmol/L. The results revealed that compounds2,3and their isomers showed moderate inhibitory activity against Top1 at 50mmol/L (Fig. 2A and B). Moreover,the (S)-isomers exhibited higher inhibitory activity than the (R)-isomers. As shown in Fig. 2C,Compounds 2,3and four isomers showed moderate to good inhibitory activity against Top2. Most of them were more potent than Eto at 50mmol/L. Similarly,the (S)-isomers exhibited stronger activity than the (R)-isomers. It is concluded that the (S)-isomers are more active than the (R)-isomers. This result was consistent with that obtained from the cytotoxicity assay.

|
Download:
|
| Fig. 2. Top1 and Top2 inhibitory activity of compounds. (A) Inhibition of Top1 relaxation activity at 50mmol/L. Lane 1,supercoiled plasmid DNA; Lane 2,DNA + Top1; Lanes 3, DNA + Top1 + CPT; Lanes 4-6,DNA + Top1 + evodiamine derivatives (3,((S)-3,((R)-3); (B) Lane 1,DNA + Top1; Lanes 2,DNA + Top1 + CPT; Lanes 3-5,DNA + Top1 + evodiamine derivatives (2,((S)-2,((R)-2). (C) Inhibition of Top2 relaxation activity at 50mmol/L. Lane 1,supercoiled plasmid DNA; Lane 2,DNA + Top2; Lanes 3,DNA + Top2 + Eto; Lanes 4- 9,DNA + Top2 + evodiamine derivatives (2,((S)-2,((R)-2,3,((S)-3,((R)-3). | |
{kind=link}
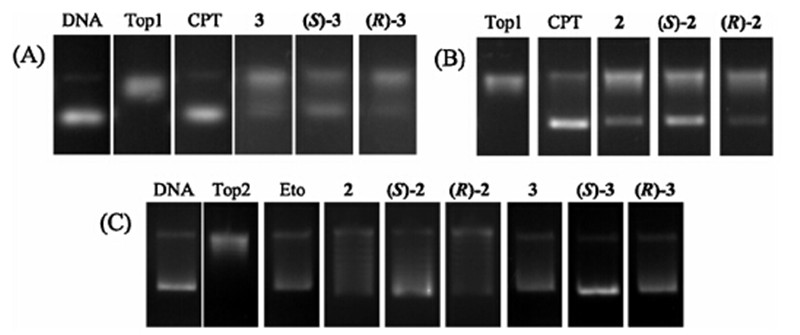
Molecular docking was performed to evaluate the binding mode of (S)-2with Topoisomerase I and II and the result was shown in Fig. 3. As indicated in the interaction model between (S)-2and Top1-DNA complex (Fig. 3A),the binding mode was very similar with evodiamine. The ABC-ring intercalates at the site of DNA cleavage and formp-stacking interactions with both the-1 (upstream) and +1 (downstream) base pairs. The N atom of pyridine forms a hydrogen bond with the Arg364. DE-ring is faced into the minor groove. The binding mode of (S)-2with Top2 was also investigated (Fig. 3B). The D-ring oxygen atom undergoes a hydrogen bond with Asn150. Moreover,the E-ring showed an effective cation-pinteraction with the magnesium cation.

|
Download:
|
| Fig. 3. Binding mode of (S)-2with Top1/Top2. (A) Binding mode of (S)-2to Top1-DNA complex. (B) Binding mode of (S)-2to the ATP-binding domain of Top2. The figure was generated using PyMol (http://www.pymol.org/). | |
{kind=link}
In this study,isomers of evodiamine derivatives 2 and3were obtained by straightforward asymmetric total synthesis and their antitumor activity was investigated. All the four isomers exhibited good to excellent antitumor potency. The Top1/Top2 inhibitory assay and further molecular docking confirmed that the isomers acted by dual inhibition of Top1 and Top2. The (S)-isomers were more active than the (R)-isomers against Top1/Top2. This study provided a vital data for further research of the isomers of evodiamine derivatives.
AcknowledgmentThis work was supported by National Natural Science Foundation of China (Nos. 81222044,81373278) and Key Project of Science and Technology of Shanghai (Nos. 11431920402, 14YF1405400).
| [1] | G.M. Cragg, P.G. Grothaus, D.J. Newman, Impact of natural products on developing new anti-cancer agents, Chem. Rev. 109 (2009) 3012-3043. |
| [2] | D.J. Newman, G.M. Cragg, Natural products as sources of new drugs over the 30 years from 1981 to 2010, J. Nat. Prod. 75 (2012) 311-335. |
| [3] | G.M. Cragg, D.J. Newman, Natural products: a continuing source of novel drug leads, Biochim. Biophys. Acta 1830 (2013) 3670-3695. |
| [4] | J. Jiang, C. Hu, Evodiamine: a novel anti-cancer alkaloid from Evodia rutaecarpa, Molecules 14 (2009) 1852-1859. |
| [5] | H. Yu, H. Jin, W. Gong, et al., Pharmacological actions of multi-target-directed evodiamine, Molecules 18 (2013) 1826-1843. |
| [6] | G. Dong, C. Sheng, S. Wang, et al., Selection of evodiamine as a novel topoisomerase I inhibitor by structure-based virtual screening and hit optimization of evodiamine derivatives as antitumor agents, J. Med. Chem. 53 (2010) 7521-7531. |
| [7] | G. Dong, S. Wang, Z. Miao, et al., New tricks for an old natural product: discovery of highly potent evodiamine derivatives as novel antitumor agents by systemic structure-activity relationship analysis and biological evaluations, J. Med. Chem. 55 (2012) 7593-7613. |
| [8] | K. Mori, Bioactive natural products and chirality, Chirality 23 (2011) 449-462. |
| [9] | M. Decker, Novel inhibitors of acetyl-and butyrylcholinesterase derived from the alkaloids dehydroevodiamine and rutaecarpine, Eur. J. Med. Chem. 40 (2005) 305-313. |
| [10] | A. Nakayama, N. Kogure, M. kitajima, et al., Straightforward asymmetric total synthesis of (+)-evodiamine, a major indole alkloid in herbal medicine "Wu Zhu Yu", Heterocycles 76 (2008) 861-865. |
| [11] | H. Huang, Q. Chen, X. Ku, et al., A series of alpha-heterocyclic carboxaldehyde thiosemicarbazones inhibit topoisomerase IIalpha catalytic activity, J. Med. Chem. 53 (2010) 3048-3064. |
| [12] | L. Du, H.C. Liu, W. Fu, et al., Unprecedented citrinin trimer tricitinol B functions as a novel topoisomerase IIalpha inhibitor, J. Med. Chem. 54 (2011) 5796-5810. |
| [13] | B.L. Staker, M.D. Feese, M. Cushman, et al., Structures of three classes of anticancer agents bound to the human topoisomerase I-DNA covalent complex, J. Med. Chem. 48 (2005) 2336-2345. |
| [14] | H. Wei, A.J. Ruthenburg, S.K. Bechis, et al., Nucleotide-dependent domain movement in the ATPase domain of a human type IIA DNA topoisomerase, J. Biol. Chem. 280 (2005) 37041-37047. |
| [15] | Accelrys Software Inc., Discovery Studio Modeling Environment, Release 3.0, Accelrys Software Inc., San Diego, 2010. |
| [16] | G. Jones, P. Willett, R.C. Glen, et al., Development and validation of a genetic algorithm for flexible docking, J. Mol. Biol. 267 (1997) 727-748. |
| [17] | K. Fang, G. Dong, H. Gong, et al., Design, synthesis and biological evaluation of E-ring modified evodiamine derivatives as novel antitumor agents, Chin. Chem. Lett. 25 (2014) 978-982. |