



 , Xiao-Dong Maa
, Xiao-Dong Maa
b Key Laboratory of Animal Models and Human Disease Mechanisms of Chinese Academy of Sciences and Yunnan Province, Kunming Institute of Zoology, Chinese Academy of Sciences, Kunming 650223, China;
c Kunming College of Life Science, University of Chinese Academy of Sciences, Kunming 650204, China
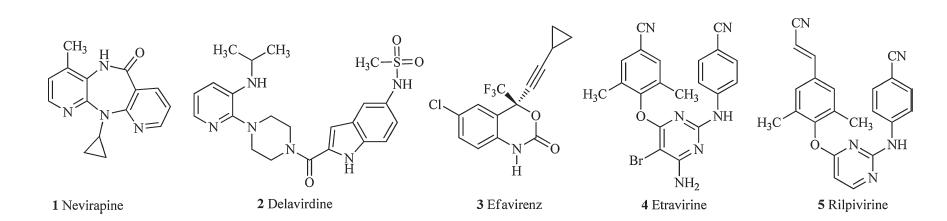
Non-nucleoside reverse transcriptase inhibitors (NNRTIs) are important part of certain multidrug regimens used in the treatment of HIV infection [1, 2, 3, 4]. Till now,five NNRTIs (Fig. 1): nevirapine (1,NVP) [5],delavirdine (2,DLV) [6],efavirenz (3,EFE) [7] etravirine (4,ETV) [8],and rilpivirine (5,RPV) [9, 10, 11] have been approved for the clinical use,and significantly reduced the deaths from AIDS. However,for the rapid emergence of drug resistance, more effective NNRTIs,in particular those with new chemotypes are still required in order to allow continued suppression of the viral infection [12, 13].

|
Download:
|
| Fig. 1. Structures of currently marketed NNRTIs. | |
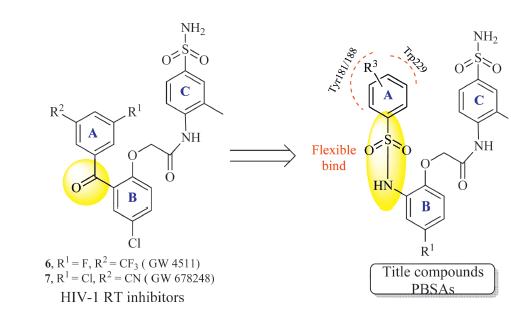
Recently,the novel benzophenone derivatives aroused great attention due to their broad spectrum activity against several key mutant HIV-1 strains,such as Tyr188,Tyr181,Trp229,and so on [14, 15, 16, 17]. Of these compounds,GW4511 (6,Fig. 2) [14] and GW678248 (7,Fig. 2) [15, 16] were the most active inhibitors,and dramatically improved the drug resistance issues. In particular, GW678248,which was progressed to the clinical II studies,also possesses excellent pharmacokinetic property. As our great interest, we have carried out some modifications on this scaffold,and obtained Ami-BPs [18],Naph-BPs [19],Eth-BPs [20],and Hyd-BPs [21] as newpotent BP inhibitors. Previous SAR explorations revealed that the flexible linker between A- and B-rings could not only improve the adaptation of the inhibitor to RT,but also promote the H-bond bindings with Tyr188 or Tyr181 [18]. To further enhance these actions,we designed a series of phenylbenzenesulfonamide derivatives (PBSAs,Fig. 2),in which the A-ring was more flexible. In this manuscript,these compoundswere synthesized,and evaluated for their preliminary biological activity against wild-type HIV-1 virus.Moreover,applying themolecular stimulations,the actions of the potent inhibitor 13f with HIV-1 RT was also investigated.

|
Download:
|
| Fig. 2. Designing strategy of the title compounds. | |
All chemicals were purchased from commercial sources, analytical grade and used without further purification. Melting points were measured on a SGW X-4 microscopic melting-point apparatus and were uncorrected. Mass spectra were obtained on a Waters Quattro Micromass instrument using electrospray ionization (ESI) techniques. 1H NMR and 13C NMR spectra on a Brucker AV 400 MHz spectrometer were recorded in DMSO-d6. Chemical shifts were reported in d (ppm) units relative to the internal standard tetramethylsilane (TMS). Elemental analyses were performed on a Carlo Erba 1106 instrument and the results of elemental analyses for C,H,Cl,Br,F and N were within ±0.4% of the theoretical values.
Applying our previously reported procedure [18],all these newly designed compounds were first synthesized. The following are general procedure and structure datas of typical inhibitors. A mixture of 12 (0.01 mol) and pyridine (3.36 g,0.04 mol) in acetone (50 mL) was cooled to 0 8C in an ice bath,then aryl sulfonyl chloride (0.02 mol) was added dropwise slowly. The resulting mixture was allowed to warm to room temperature and stirred for another 10 min. After the reaction was complete,the mixture was poured into water (150 mL),filtered and washed with ethyl ester to give crude product as beige solid,which was purified by crystallization from acetonitrile to provide the target compounds 13a-t as off-white solids.
N-[4-(Aminosulfonyl)-2-methylphenyl]-2-[4-chloro-2-(4- methylbenzenesulfonyl)aminophenoxy]acetamide (13a): Yield 66.1%; off-white solid,mp 132.2-134.1 8C; 1H NMR (DMSO-d6): δ 2.22 (s,3H,CH3),2.31 (s,3H,CH3),4.35 (s,2H,CH2),7.32 (s,2H,NH2), 6.95-7.82(m,10H,PhH),9.57 (br,1H,NH),9.71 (s,1H,NH); 13CNMR (DMSO-d6): δ 18.5,22.3,68.1,114.1,121.7,122.8,125.9,126.1, 126.4,126.8,127.2 (2C),128.2,130.1 (2C),134.2,137.3,139.9,142.1, 144.4,150.1,166.8; MS (ESI-) m/z 523 [M-H]-; Anal. Calcd. for C22H22ClN3O6S2: C 50.43,H 4.23,Cl 6.77,N 8.02,O 18.32,S 12.24, found: C 50.47,H 4.21,Cl 6.76,N 8.01,O 18.30,S 12.26.
N-[4-(Aminosulfonyl)phenyl]-2-[4-chloro-2-(4-methylbenzenesulfonyl) aminophenoxy]acetamide (13f): Yield 71.1%; off-white solid,mp 201.1-202.9 8C; 1H NMR (DMSO-d6): δ 2.27 (s,3H,CH3), 4.26 (s,2H,CH2),7.33(s,2H,NH2),6.67-7.93 (m,11H,PhH),9.59 (s, 1H,NH),9.88 (br,1H,NH); 13C NMR (DMSO-d6): δ 18.5,68.1,113.5, 121.9 (2C),122.3,126.1,126.3,127.1(2C),127.5 (2C),128.1,129.3 (2C),135.1,138.6,140.3,142.8,151.3,166.4; MS (ESI-) m/z 509 [M-H]-; Anal. Calcd. for C21H20ClN3O6S2: C 49.46,H 3.95, Cl 6.95,N 8.24,O 18.82,S 12.57,found: C 49.50,H3.93,Cl 6.93,N 8.23, O 18.81,S 12.59.
N-[4-(Aminosulfonyl)phenyl]-2-[4-chloro-2-(4-methoxybenzenesulfonyl) aminophenoxy]acetamide (13j): Yield 53.9%; white solid,mp 243.1-244.6 8C; 1H NMR (DMSO-d6): δ 3.68 (s,3H,OCH3), 4.35 (s,2H,CH2),7.35 (s,2H,NH2),6.85-7.80 (m,11H,PhH),9.57 (s, 1H,NH),9.91 (s,1H,NH); 13C NMR (DMSO-d6): δ 55.9,58.3,114.2, 115.3 (2C),121.1 (2C),123.2,125.9,126.7,127.9 (2C),128.3,127.0 (2C),133.4,140.6,142.5,152.9,162.5,167.8; MS (ESI-) m/z 525 [M-H]-; Anal. Calcd. for C21H20ClN3O7S2: C 47.95,H 3.83,Cl 6.74,N7.99,O 21.29,S 12.19,found: C 47.98,H3.82,Cl 6.72,N 7.97, O 21.30,S 12.20.
N-[4-(Aminosulfonyl)-2-methylphenyl]-2-[2-(4-methylbenzenesulfonyl) aminophenoxy]acetamide (13k): Yield 56.4%; white solid,mp 119.0-120.1 8C; 1H NMR (DMSO-d6): δ 2.25 (s,3H,CH3), 2.32 (s,3H,CH3),4.37 (s,2H,CH2),7.33 (s,2H,NH2),6.97-7.75 (m, 11H,PhH),9.65 (br,1H,NH),9.73 (s,1H,NH); 13C NMR (DMSO-d6): d 18.7,21.8,68.5,114.3,122.7,124.8,126.4,126.1,126.8,126.9, 127.5(2C),128.6,130.2(2C),134.5,138.1,139.2,142.4,144.0, 150.8,167.2; MS (ESI-) m/z 488 [M-H]-; Anal. Calcd. for C22H23N3O6S2: C 53.97,H 4.74,N 8.58,O 19.61,S 13.10,found: C 53.94,H 4.73,N 8.59,O 19.60,S 13.14.
N-[4-(Aminosulfonyl)phenyl]-2-[2-(4-chlorobenzenesulfonyl)- aminophenoxy] acetamide (13t): Yield 68.1%; off-white solid,mp 137.7-138.9 8C; 1H NMR(DMSO-d6): δ 4.38 (s,2H,CH2),7.30 (s,2H, NH2),6.70-7.87 (m,12H,PhH),9.87 (s,1H,NH),10.04 (br,1H,NH); 13C NMR (DMSO-d6): δ 68.8,114.7,120.8 (2C),122.9,127.2,127.5, 127.6(2C),127.8,129.3 (2C),129.8 (2C),134.4,138.3,140.2,141.7, 151.5,167.5; MS (ESI-) m/z 495 [M-H]-; Anal. Calcd. for C20H18ClN3O6S2: C 48.43,H 3.66,Cl 7.15,N 8.47,O 19.36,S 12.93,found: C 48.40,H 3.63,Cl 7.17,N 8.46,O 19.40,S 12.94.
Cytotoxicity assay: The cytotoxicities of compounds on C8166 cells were assessed by MTT colorimetric assay as described previously [22]. The absorbance at 570 nm/630 nm (A570/630) was read in an ELISA reader (Elx1000,Bio-Tek Instrument Inc.,USA). The minimumcytotoxic concentration that caused the reductionof viable cells by 50% (CC50) was determined from the dose response curve.
Syncytium reduction assay: In the presence of 100 μL of various concentrations of compounds,C8166 cells (4 × 105/mL) were infected with virus (HIV-1 IIIB) at a multiplicity of infection (M.O.I) of 0.06. The final volume per well was 200 μL. AZT and GW678248 were used for drug control. After 3 days of culture,the number of syncytia (multinucleated giant cells) was scored under an inverted microscope; 50% effective concentration to blocking syncytia formation (EC50) was calculated [22]. 3. Results and discussion
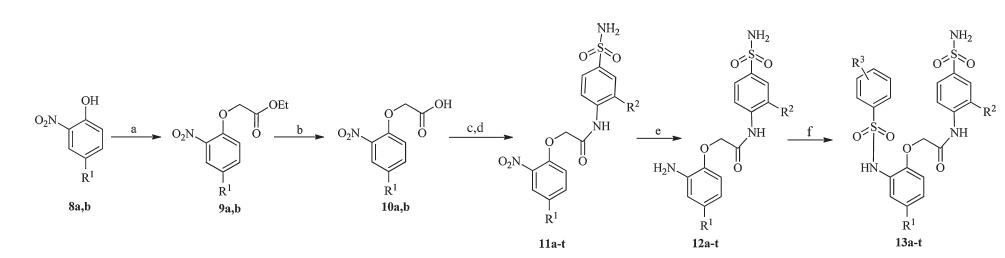
According to the established methodology [18],treatment of 2-nitrophenol (8a,b) with ethyl bromoacetate,and subsequent hydrolysis provides the acid derivative 10a,b. After chlorination of 10,and then coupling with 4-amino-benzenesulfonamide in basic condition,intermediates 11a-t were prepared in good yields (66%-86%). Next,reducing reaction in the presence of Fe-NH4Cl was carried out to obtain the amino 12a-t. Then by coupling 12 with the appropriately substituted of aryl sulfonyl chloride under the action of base,the title compounds 13a-t were furnished in 53%-74% yields (Scheme 1).

|
Download:
|
| Scheme 1. Synthesis of N-phenylbenzenesulfonamide analogues 13a-t. Reagents and conditions: (a) BrCH2COOEt,K2CO3,KI,acetonitrile,2 h,80 8C,72%-90%; (b) NaOH,H2O, 60 8C,2 h,68%-84%; (c) SOCl2,80 8C,1 h; (d) 4-aminobenzenesulfonamide,NaHCO3,acetone,80 8C,2 h,66%-86%; (e) Fe,NH4Cl,acetone-H2O,80 8C,12 h,58%-76%; (f) RSO2Cl,Py,acetonitrile,r.t,30 min,53%-74%. | |
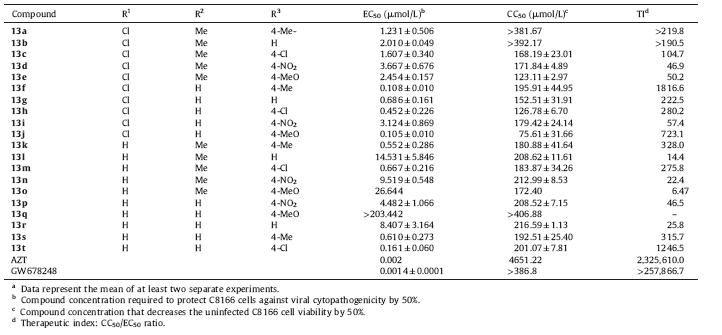
To explore the biological activity of these newly obtained analogues,the activity against wild-type HIV-1 strain IIIB was tested [22]. For comparison,lead compound GW678248 and the FDA-approved drug zidovudine (AZT) were also evaluated as reference compounds. The activity data is interpreted in CC50 (cytotoxicity),EC50 (anti-HIV activity) and TI (therapeutic index, given by the CC50/EC50 ratio).
The assay results for 13a-t are summarized in Table 1, apparently,most of these compounds exhibit moderate activity against the wild-type HIV-1 strain IIIB with EC50 values ranging from 0.105 mmol/L to 14.531 μmol/L,along with the TI values ranging from 12.90 to 1816.55. The most active inhibitor 13f with a TI value of 1816.55 could able to inhibit the wild-type HIV-1 at very low concentrations with an EC50 value of 0.108 μmol/L. It is no doubt that the PBSA derivatives were able to form binding affinity with HIV-1 RT. However,the actions were so weak that they conferred an anti-HIV activity about 100-fold lower than GW678248.
| Table 1 Biological activity of compounds 13a-t against HIV-1 in C8166 cells.a |
{kind=link}
{kind=link}
{kind=link}
By placing various substituents on the A-,B-,and C-rings,a series of PBSAs were synthesized to explore their SARs. For the benzenesulfonamide section,which closed to the top hydrophobic pocket lined by the important aromatic residues Tyr181,Tyr188 and Trp229,a number of C-4 substituents (H,Me,NO2,Cl,MeO) were introduced (compounds 13a-e). Among 13a-e,most of them have similar level of anti-HIV activity (1.607-3.667 μmol/L),as well as the higher cytopathogenicity (>123.11 μmol/L). Interestingly, just removing the methyl group at C-2 position of the C-ring, the inhibition abilities of analogues 13f-i (<1 μmol/L) were tremendously improved. The notable example was compound 13f (0.108 μmol/L),which almost displayed 20 times stronger potency compared with 13b (2.010 μmol/L). Another series of compounds 13k-o,with the removal of the chlorine atom on the B-ring,also exhibit weak anti-HIV activity,except for analogues 13k and m (0.552 μmol/L and 0.667 μmol/L,respectively). Furthermore, compounds 13p-t were also synthesized to explore the affect of the methyl group on the C-ring. Apparently,this modification was more favourable,and resulted in the identification of a very potent compound 13t,exhibiting 0.161 μmol/L anti-wild-type HV-1 activity and 1246.50 selectivity.
Overall,our previous SAR studies implied that installing substituents on the B-ring would decrease the anti-HIV activity. In terms of B- and C-rings,more substituents were needed to establish their SARs. Although the obtained PBSAs were not as potent as GW678248,they provided a new potentia anti-HIV template for further optimizations.
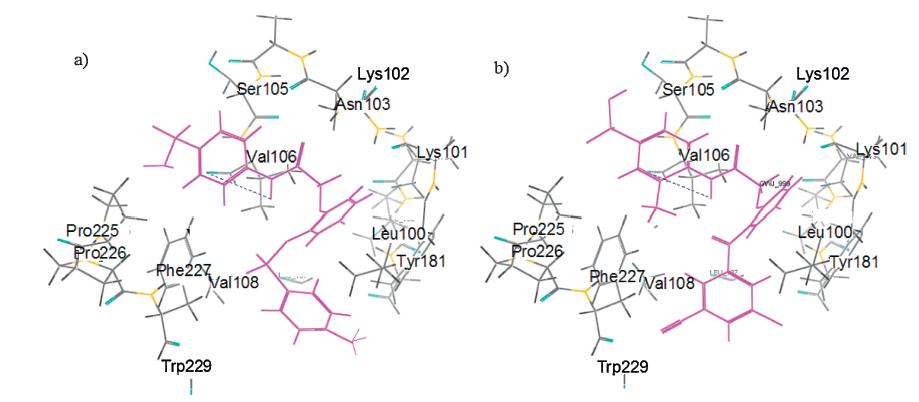
In addition,using the program AutoDock 4.0.1 [23],a modelling study was performed to explore the possible action mechanism of the PBSAs with HIV-1 RT. According to the basis of the protocols, the most active inhibitor 13f was chosen to be docked into the nonnucleoside inhibitor binding pocket (NNIBP) of RT (PDB code: 3DOK). The default AutoDock 4.0.1 parameters were used. For comparison,GW678248 was also docked to NNIBP with the same protocols.
It was clear to indicate that the binding conformations of compound 13f and GW678248 were very similar,especially in the hydrophilic section (Fig. 3). Of this template,the sulfonamide group on the C-ring reached the top of the cavity and formed strong actions with Ser105,Val105,Val106 and PrO225,while the NH part that linked the B- and C-rings formed a direct hydrogen bond with the backbone OH group of Asn103,in which the oxygen atom serves as hydrogen-bond acceptor (average O. . .H distance = 2.938A˚ ). At the hydrophobic part,the aromatic ring A in GW678248 pointed to the top of the cavity and interacted with several key residues,such as Trp229,Val108,Phe227,Tyr181,and Leu100 through p -p and π -π stacking. Clearly,the conformation of lead compound fits nicely into the shape of the cavity. However, for the lack of groups CN,Cl,etc.,which could form strong p -p stacking interactions with Trp229 (due to the different spatial positions of the phenyl moieties,the distance between the phenyl moieties of 13f and GW678248 were 4.792 and 4.231Å , respectively),analogue 17f significantly lost its potency in repressing HIV virus.

|
Download:
|
| Fig. 3. (a) Model of 13f docked into the non-nucleoside binding site of RT; (b) Model of GW678248 docked into the non-nucleoside binding site of RT. | |
{kind=link}
In summary,being a novel anti-HIV template,N-phenylbenzenesulfonamide derivatives demonstrate potential anti-wild-type activity with EC50 values ranging from 0.105 mmol/L to 14.531 mmol/L. In particular,compound 13f not only has high anti-HIV activity (0.108 μmol/L),but also possesses low toxicity (TI = 1816.6). Additionally,computer stimulation also disclosed that long distance between A-ring and HIV-1 RT BBINP was the main reason for their dropped activity. To develop more active PBSAs as HIV inhibitors,various substitutes installed in the A-ring will be beneficial.
AcknowledgmentsThis work was supported in part by grants from the National Natural Science Foundation of China (No. 81402788), and the Ph.D. Start-up Fund of Natural Science Foundation of Liaoning Province, China (No. 20141115).
| [1] | W. Schaefer, W.G. Friebe, H. Leinert, et al., Non-nucleoside inhibitors of HIV-1 reverse transcriptase: molecular modeling and X-ray structure investigations, J. Med. Chem. 36 (1993) 726-732. |
| [2] | J.F. Palella, K.M. Delaney, A.C. Moorman, et al., Declining morbidity and mortality among patients with advanced human immunodeficiency virus infection. HIV outpatient study investigators, N. Engl. J. Med. 338 (1998) 853-860. |
| [3] | E. De Clerck, New developments in anti-HIV chemotherapy, Curr. Med. Chem. 8 (2001) 1543-1572. |
| [4] | M.P. de Be´ thune, Non-nucleoside reverse transcriptase inhibitors (NNRTIs), their discovery, development, and use in the treatment of HIV-1 infection: a review of the last 20 years (1989-2009), Antivir. Res. 85 (2010) 75-90. |
| [5] | R.A. Koup, V.J. Merluzzi, K.D. Hargrave, et al., Inhibition of human immunodeficiency virus type 1 (HIV-1) replication by the dipyridodiazepinone BI-RG-587, J. Infect. Dis. 163 (1991) 966-970. |
| [6] | W.W. Freimuth, Delavirdine mesylate, a potent non-nucleoside HIV-1 reverse transcriptase inhibitor, Adv. Exp. Med. Biol. 394 (1996) 279-289. |
| [7] | S.D. Young, S.F. Britcher, L.O. Tran, et al., L-743, 726 (DMP-266): a novel, highly potent nonnucleoside inhibitor of the human immunodeficiency virus type 1 reverse transcriptase, Antimicrob. Agents Chemother. 39 (1995) 2602-2605. |
| [8] | L.B. Johnson, L.D. Saravolatz, Etravirine, a next-generation nonnucleoside reversetranscriptase inhibitor, Clin. Infect. Dis. 48 (2009) 1123-1128. |
| [9] | S. Moreno, J. Ló pez Aldeguer, J.R. Arribas, et al., The future of antiretroviral therapy: challenges and needs, J. Antimicrob. Chemother. 65 (2010) 827-835. |
| [10] | H. Azijn, I. Tirry, J. Vingerhoets, et al., TMC278, a next-generation nonnucleoside reverse transcriptase inhibitor (NNRTI), active against wild-type and NNRTIresistant HIV-1, Antimicrob. Agents Chemother. 54 (2010) 718-727. |
| [11] | Z. Zhang, R. Hamatake, Z. Hong, Clinical utility of current NNRTIs and perspectives of new agents in this class under development, Antivir. Chem. Chemother. 15 (2004) 121-134. |
| [12] | Guidelines for the Use of Antiretro Iral Agents in HIV-1-Infected Adults and Adolescents. http://AIDSinfo.nih.gov (29.10.04). |
| [13] | R.M. Grant, F.M. Hecht, M. Warmerdam, et al., Time trends in primary HIV-1 drug resistance among recently infected persons, JAMA 288 (2002) 181-188. |
| [14] | J.H. Chan, G.A. Freeman, J.H. Tidwell, et al., Novel benzophenones as non-nucleoside reverse transcriptase inhibitors of HIV-1, J. Med. Chem. 47 (2004) 1175-1182. |
| [15] | K.R. Romines, G.A. Freeman, L.T. Schaller, et al., Structure-activity relationship studies of novel benzophenones leading to the discovery of a potent, next generation HIV nonnucleoside reverse transcriptase inhibitor, J. Med. Chem. 49 (2006) 727-739. |
| [16] | R.G. Ferris, R.J. Hazen, G.B. Roberts, et al., Antiviral activity of GW678248, a novel benzophenone nonnucleoside reverse transcriptase inhibitor, Antimicrob. Agents Chemother. 49 (2005) 4046-4051. |
| [17] | P.G. Wyatt, R.C. Bethell, N. Cammack, et al., Benzophenone derivatives: a novel series of potent and selective inhibitors of human immunodeficiency virus type 1 reverse transcriptase, J. Med. Chem. 38 (1995) 1657-1665. |
| [18] | X.D. Ma, Q.Q. He, X. Zhang, et al., Synthesis, structure-activity relationships, and docking studies of N-phenylarylformamide derivatives (PAFAs) as non-nucleoside HIV reverse transcriptase inhibitors, Eur. J. Med. Chem. 58 (2012) 504-512. |
| [19] | X.D. Ma, X. Zhang, H.F. Dai, et al., Synthesis and biological activity of naphthylsubstituted (B-ring) benzophenone derivatives as novel non-nucleoside HIV-1 reverse transcriptase inhibitors, Bioorg. Med. Chem. 19 (2011) 4601-4607. |
| [20] | X.D. Ma, X. Zhang, S.Q. Yang, et al., Synthesis and biological evaluation of (±)-benzhydrol derivatives as potent non-nucleoside HIV-1 reverse transcriptase inhibitors, Bioorg. Med. Chem. 19 (2011) 4704-4709. |
| [21] | S.X. Gu, X. Zhang, Q.Q. He, et al., Synthesis and biological evaluation of naphthyl phenyl ethers (NPEs) as novel nonnucleoside HIV-1 reverse transcriptase inhibitors, Bioorg. Med. Chem. 19 (2011) 4220-4226. |
| [22] | Y.T. Zheng, K.L. Ben, S.W. Jin, Anti-HIV-1 activity of trichobitacin, a novel ribosome-inactivating protein, Acta Pharmacol. Sin. 21 (2000) 179-182. |
| [23] | D.S. Goodsell, G.M. Morris, A.J. Olson, Automated docking of flexible ligands: applications of AutoDock, J. Mol. Recognit. 9 (1996) 1-5. |