




b Department of Chemistry, Louisiana State University, Baton Rouge, LA 70803, USA
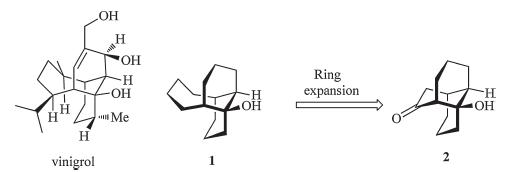
Tricyclo[6.4.0.04,9]dodecane framework represents a very unusual ring skeleton that features an unprecedented 1,5- ethano-bridged cis-decalin. Although the ring system is rarely found in biological active natural products,its homologue,with two extra CH2 groups,is represented in the famous natural product,vinigrol (Scheme 1) [1, 2, 3]. From the synthetic perspective, the skeleton of vinigrol (1) could be built by a two-carbon ring expansion strategy from the tricyclo[6.4.0.04,9]dodecane framework (2) (Scheme 1).

|
Download:
|
| Scheme 1. A ring-expansion approach to vinigrol. | |
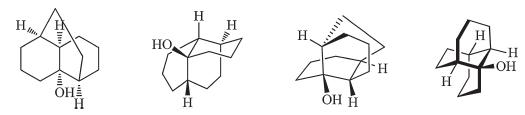
Due to the unique structural feature of the framework,it has received some attention from both synthetic [4, 5, 6, 7] and theoretical points of view [8]. Although it is relatively small in size,the presence of four contiguous stereocenters and the unusual carbon ring arrangement make it a challenging synthetic problem,which can be analyzed from several seemingly different topological viewpoints (Scheme 2). Indeed,there is only one report [4] capable of preparing the framework as a byproduct in a very low isolated yield starting from furanophane (furanophane is not commercially available,for the preparation,please see references [9, 10, 11, 12]). Herein,we disclose a general and facile synthesis of the skeleton from commercially available starting material.

|
Download:
|
| Scheme 2. Different views of tricyclo[6.4.0.04,9]dodecane framework. | |
Preparation of compound 15: To an ice-cold solution of the ketone 14 (3.0 g,14.4 mmol) in 70 mL of dry CH2Cl2 was added 2,6- lutidine (6.17 g,57.6 mmol) and TBSOTf (8.37 g,31.7 mmol) sequentially,dropwise and the resulting reaction mixture was stirred at r.t. for 3 h. Then the mixture was poured into 100 mL of saturated aqueous NaHCO3 at 0 °C. The solution was extracted thoroughly with CH2Cl2,and the organic extracts were dried over Na2SO4 and filtered. Removal of the solvent under reduced pressure followed by column with hexane as the eluent to give the doubly TBS protected product (5.6 g,89%) as colorless oil. 1H NMR (400 MHz,CDCl3): δ 5.72-5.82 (m,1H),4.97-5.06 (m,2H), 2.54-2.57 (m,1H),2.39-2.44 (m,1H),2.23-2.28 (m,1H),2.02-2.14 (m,2H),1.60-1.70 (m,2H),1.50-1.55 (m,8H),1.18-1.21 (m,1H), 0.96 (s,9H),0.87 (s,9H),0.15 (d,6H,J = 6 Hz),0.08 (d,6H,J = 7 Hz). 13C NMR (100 MHz,CDCl3): δ 142.2,138.8,115.1,114.2,74.3,48.5, 46.3,38.5,36.2,30.1,25.9,20.0,19.4,18.2,18.1,14.8,-1.8,-1.9, -3.6,-3.8. HRMS (ESI-TOF): m/z calcd. for (C25H48O2Si2+H+): 437.3266; found: 437.3260.
A solution of the above product (4.34 g,9.9 mmol) in 8.8 mL of THF was cooled to 0 °C,and a solution of 9-BBN in THF (0.5 mol/L, 19.8 mL,9.9 mmol) was added dropwise under nitrogen. The solution was slowly (about 2 h) warmed to r.t. and stirred for 3 h at r.t. (the reaction was judged complete by TLC). The reaction mixture was opened to air and NaOH solution (3 mol/L,19.8 mL, 59.4 mmol) was added. The mixture was cooled to 0 °C and H2O2 (8.5 mL,30% max.,79 mmol) was added dropwise followed by MeOH (5.6 mL). The resulting mixture was warmed to r.t. and stirred for an additional 3 h. Then the organic solvent was moved in vacuo,and the residue was diluted with ethyl acetate and washed with saturated aqueous NaHCO3 and brine. The organic layer was dried over MgSO4,filtered,concentrated and purified by silica gel chromatography (Hex/EA = 5:1) to give the product 15 (4.47 g, 99%) as colorless oil. 1H NMR (400 MHz,CDCl3): δ 3.60-3.69 (m, 2H),2.40-2.41 (m,1H),2.33-2.37 (m,2H),1.75-1.80 (m,4H), 1.40-1.54 (m,10H),0.96 (s,9H),0.86 (s,9H),0.14 (d,6H,J = 6 Hz), 0.07 (d,6H,J = 6 Hz). 13C NMR (100 MHz,CDCl3): δ 142.4,142.3, 114.0,109.8,75.2,74.6,63.5,49.1,48.5,46.5,43.5,43.2,41.0,39.3, 36.2,31.5,28.4,25.9,22.8,21.2,20.3,19.3,18.2,15.2,14.9,-1.8, -3.7. HRMS (ESI-TOF): m/z calcd. for (C25H50O3Si2+H+): 455.3371; found: 455.3360.
Preparation of compound 16: To a solution of the alcohol 15 (4.47 g,9.8 mmol) in 65 mL of THF at -78 °C was added a solution of TBAF (1.0 mol/L,10 mL,10.0 mmol) in THF. Then the reaction mixture was increased to 0 °C and stirred for 1.5 h,and then quenched with water. The mixture was then extracted with ethyl acetate and the combined organic phases were washed with brine, dried (MgSO4),filtered and concentrated. Column chromatography (Hex/EA = 3:1) afforded the product 16 (2.8 g,84%) as colorless oil. 1H NMR (400 MHz,CDCl3): δ 3.70 (t,2H,J = 5 Hz),2.64-2.68 (m, 2H),2.40-2.58 (m,2H),2.37-2.38 (m,1H),1.85-1.87 (m,2H), 1.73-1.76 (m,2H),1.45-1.56 (m,5H),1.10-1.30 (m,1H),1.08 (d, 3H,J = 7 Hz),0.86 (s,9H),0.08 (s,6H). 13C NMR (100 MHz,CDCl3): δ 212.4,76.0,63.0,57.0,50.0,48.9,41.0,38.8,34.8,31.3,27.3,25.7, 21.9,19.4,18.8,18.0,11.8,-1.9. HRMS (ESI-TOF): m/z calcd. for (C19H36O3Si+Na+): 363.2326; found: 363.2332.
Preparation of compound 17: To a stirred solution of the ketone 14 (1.23 g,5.9 mmol) in 100 mL of dichloromethane/diisopropyl ethylamine (1:1) at 0 °C was added dropwise MOMCl (4.5 mL, 59 mmol). After stirring 24 h at r.t.,the reaction mixture was diluted with 50 mL of DCM and the organic layer was washed with 50 mL of 3 mol/L HCl. The aqueous washes were back extracted with DCM (3× 50 mL),and the combined organic layers were washed with brine,dried (MgSO4),filtered and concentrated. The residue was chromatographed on silica gel (Hex/EA = 10:1) yielded the titled compound 17 (1.08 g,73%) as pale yellow oil. 1H NMR (400 MHz,CDCl3): δ 5.76-5.83 (m,1H),5.05-5.12 (m,2H),4.79 (d, 1H,J = 7.0 Hz),4.70 (d,1H,J = 7.0 Hz),3.37 (s,3H),2.71-2.79 (m, 1H),2.60-2.68 (m,2H),2.36-2.39 (m,1H),2.09-2.22 (m,2H), 1.48-1.75 (m,6H),1.20-1.33(m,1H),1.08 (d,3H,J = 7.0 Hz). 13C NMR (100 MHz,CDCl3): δ 211.9,137.2,116.5,89.9,78.2,55.4,53.3, 48.7,46.6,38.9,30.8,21.9,20.0,18.9,13.2. HRMS (ESI-TOF): m/z calcd. for (C15H24O3+Na+): 275.1618; found: 275.1630.
Preparation of compound 18: Borane-tetrahydrofuran (1.0mol/L in THF,0.46 mL,0.46mmol) was placed in a dry,nitrogen-flushed, round-bottomed flask,which was then immersed in an ice-water bath. Cyclohexene (0.093mL,0.92mmol) was added dropwise and the mixture stirred at 0 °C for 1 h. The substrate 17 (116 mg, 0.46mmol) in 0.3 mL of THF was then added to the slurry of dicyclohexylborane in THF. The cooling bath was removed and the mixture stirred for two hours at r.t. Oxidation was achieved by adding NaBO3·4H2O (0.212 g,1.38mmol) and water (0.5 mL) and stirring was continued at r.t. for additional 2 h. The product was extracted into ether (3× 3mL),dried (MgSO4),filtered and concentrated. The residue was chromatographed on silica gel (Hex/EA = 1:1) yielding the title compound 18 (120 mg,97%) as colorless oil. 1H NMR (250 MHz,CDCl3): δ 4.70 (d,1H,J= 7.0 Hz), 4.64 (d,1H,J= 7.0 Hz),3.61 (t,2H,J= 6.0 Hz),3.29 (s,3H),2.56-2.65 (m,3H),2.32-2.34 (m,1H),2.12 (br s,1H),1.80-2.00 (m,1H),1.63- 1.79 (m,4H),1.41-1.45 (m,4H),1.15-1.19 (m,1H),1.01 (d,3H, J= 6.8 Hz). 13CNMR(62.5MHz,CDCl3): d211.9,89.9,78.2,62.5,55.2, 53.4,48.7,46.6,38.9,31.4,30.8,21.9,19.0,18.6,12.1. HRMS (ESI-TOF): m/z calcd. for (C15H26O4+Na+): 293.1723; found: 293.1730.
Preparation of compound 19: To dry DCM (28 mL) was added in this order: triphenylphosphine (2.18 g,8 mmol),imidazole (0.56 g, 8 mmol) and iodine (2.11 g,8 mmol). A solution of the alcohol 16 (2.35 g,7 mmol) in 7 mL of dry DCM was added and the mixture was stirred at room temperature for 2 h. Then the solvent was removed in vacuo and the product was purified by passing it through a column (Hex/EA = 20:1) to afford the product 19 (2.9 g, 93%) as pale yellow crystalline solid. 1H NMR (400 MHz,CDCl3): δ 3.20-3.26 (m,2H),2.65-2.68 (m,1H),2.34-2.49 (m,2H),2.11-2.15 (m,1H),1.96-1.98 (m,2H),1.73-1.85 (m,3H),1.46-1.56 (m,4H), 1.22-1.24 (m,1H),1.05 (d,3H,J= 7 Hz),0.88 (s,9H),0.09 (s,6H). 13C NMR (100 MHz,CDCl3): δ 211.9,76.5,51.4,49.2,42.6,41.1, 39.1,32.0,27.3,27.3,25.9,20.1,19.0,11.9,7.0,-1.8. HRMS (ESITOF): m/z calcd. for (C19H35IO2Si+H+): 451.1524; found: 451.1499.
Preparation of compound 20: The alcohol 18 (94 mg, 0.35 mmol) was dissolved in THF (2.3 mL) and treated with imidazole (59.2 mg,0.87 mmol) and triphenylphosphine (109.4 mg,0.42 mmol). The mixture was cooled to 0 °C and iodine (106 mg,0.42 mmol) was added in two portions (or added portionwise over 10 min). The reaction mixture was stirred at 0 °C for 2.5 h,warmed to ambient temperature and stirred for another 1 h. The reaction mixture was quenched with saturated sodium thiosulphate (0.16 mL) and water (0.33 mL) and then stirred for 30 min at ambient temperature,poured into water (1.6 mL) and extracted with ethyl acetate (2 × 3.2 mL). The combined organic layers were dried with MgSO4,filtered and concentrated in vacuo. The residue was chromatographed on silica gel (Hex/EA = 5:1) to yield the titled compound 20 (126 mg,95%) as colorless oil. 1H NMR (400 MHz,CDCl3): δ 4.76 (d,1H, J= 7.50 Hz),4.70 (d,1H,J= 7.50 Hz),3.36 (s,3H),3.19-3.29 (m, 2H),2.76 (d,1H,J= 15.0 Hz),2.65 (d,1H,J= 15.0 Hz),2.37-2.40 (m, 1H),2.14 (br s,1H),1.86-2.02 (m,4H),1.72-1.74 (m,2H),1.48- 1.54 (m,4H),1.21-1.24 (m,1H),1.08 (d,3H,J= 6.8 Hz). 13C NMR (100 MHz,CDCl3): δ 211.6,90.2,78.0,55.4,53.6,48.8,46.6,39.2, 32.3,31.6,27.3,19.3,18.8,12.3,7.1. HRMS (ESI-TOF):m/z calcd. for (C15H25IO3+Na+): 403.0741; found: 403.0737.
Preparation of compound 21: To a solution of dry TMP (2,2,6,6- tetramethylpiperidine,1.14 mL,6.56 mmol) in 7.0 mL of THF was added n-butyllithium solution (1.6 mol/L,4.1 mL,6.56 mmol) slowly at -78 °C under N2 and increased the temperature to 0 °C. After 15 min stirring at 0 °C,the mixture was cooled to-78 °C. A solution of the substrate 19 (1.97 g,4.37 mmol) in 27 mL of THF was added dropwise to the mixture at -78 °C,and the resulting mixture was slowly warmed to -15 °C during the next 1 h, and stirring was continued overnight. Saturated aqueous NH4Cl (10 mL) was added and diluted with 50 mL of ether, separated. The organic layer was washed with water (2 × 8 mL), brine (10 mL),dried over MgSO4,filtered and concentrated under reduced pressure. The crude product NMR indicated that the ratio of two isomers is around 3.8:1. The residue was purified by column (Hex:EA = 30:1) to give the unreactive isomer 19b (0.80 g) and the desired tricyclic product 21 (0.56 g,40%) as white crystalline solid. 1H NMR (400 MHz,CDCl3): δ 2.64-2.70 (m,1H),2.40-2.47 (m,3H), 2.19-2.39 (m,1H),1.78-1.83 (m,3H),1.58-1.76 (m,1H),1.52-1.56 (m,5H),1.38-1.42 (m,1H),1.23-1.30 (m,1H),1.07 (d,3H,J= 7 Hz), 0.92 (s,9H),0.11 (d,6H,J= 7 Hz). 13C NMR (100 MHz,CDCl3): δ 216.8,75.1,57.9,45.5,44.1,42.3,41.5,28.0,26.1,25.6,24.4,20.8, 19.6,18.6,12.4,-1.6,-1.8. HRMS (ESI-TOF): m/z calcd. for (C19H34O2Si+H+): 323.2401; found: 323.2396.
Preparation of compound 23: A procedure analogous to that used for 21 afforded the unreactive isomer 20b (1.9 g) and the desired product 23 (0.9 g,27% as using 5.0 g of the SM) as colorless oil. 1H NMR (400 MHz,CDCl3): δ 4.70 (d,1H,J= 6.80 Hz),4.68 (d, 1H,J= 6.80 Hz),3.35 (s,3H),2.69 (t,1H,J= 6.8 Hz),2.60 (br s,1H), 2.20-2.28 (m,3H),1.92 (br s,1H),1.70-1.81 (m,3H),1.48-1.59 (m, 5H),1.35-1.39 (m,1H),1.19-1.28 (m,1H),1.04 (d,3H,J= 6.6 Hz). 13C NMR (100 MHz,CDCl3): δ 215.8,89.8,76.8,55.5,54.9,45.7, 42.4,41.3,36.6,27.8,25.4,24.1,19.1,18.7,12.4. HRMS (ESI-TOF): m/z calcd. for (C15H24O3+H+): 253.1798; found: 253.1789. 3. Results and discussion
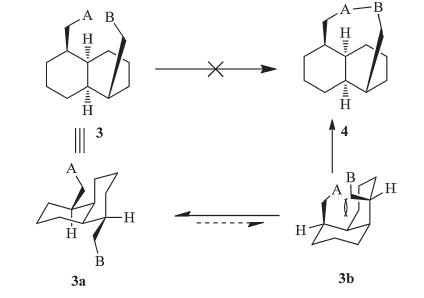
Although cis-decalin appears to be a core part of the tricyclo[6.4.0.04,9]dodecane framework 4,it is very difficult to form the bridging six-membered ring of 4 from a pre-existing cisdecalin framework 3 (Scheme 3). The major conformer 3a will adopt a diequitorial conformation,lacking the proximity needed for ring closure. Although the diaxial conformer 3b is reactive,it is too unstable to exist. Indeed,the similar problem was encountered in the synthesis of the skeleton of vinigrol. A variety of approaches from Paquette’s group have demonstrated the unlikely formation of the bridging eight-membered ring of vinigrol from a pre-existing cis-decalin framework [13, 14, 15, 16].

|
Download:
|
| Scheme 3. The intrinsic challenge approach to 1,5-bridged cis-decalin from a preexisting cis-decalin. | |
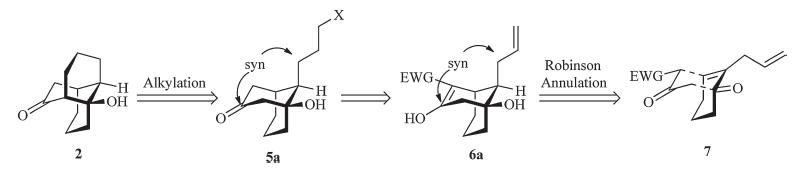
In order to avoid the cis-decalin framework in approaching the bridging six-membered ring of the tricyclic framework,we envisioned that the bridging six-membered ring of 2 could be constructed by an intramolecular alkylation from the bicyclo[ 3.3.1]nonane 5a (Scheme 4). According to Baldwin’s rule,such a 6-exo-tet cyclization should prove favorable [17, 18]. 5a could be prepared by functional group manipulation from 6a,which would be readily accessed by a Robinson annulation depicted schematically in structure 7. Obviously,the stereochemistry of the allyl group attached to the one-carbon bridge formed in the reaction is critical to the key ring closure reaction,and this diastereoselection of the reaction has been investigated carefully in our previous publication [19].

|
Download:
|
| Scheme 4. Retrosynthetic analysis of the tricyclo[6.4.0.04,9]dodecane framework. | |
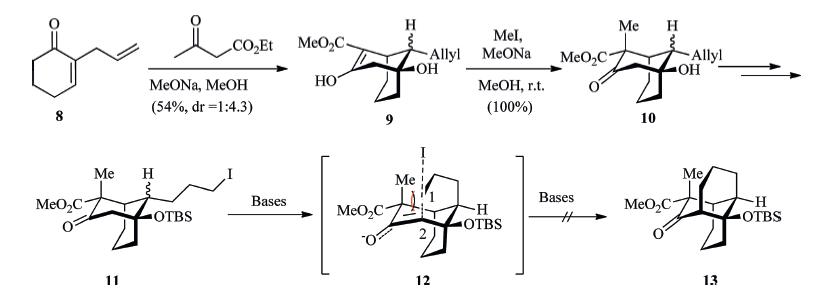
Please refer to [19] for the detailed preparation of compound 10. After several steps transformation,the cyclization precursor 11 was obtained (Scheme 5). Having iodide 11 in hand,we turned our attention to the intramolecular alkylation [20]. Unfortunately,our preliminary attempts at the preparation of the tricyclic 13 were fruitless,even though various reaction conditions were explored. Although the iodide 11 was a mixture of diastereomers with the unreactive isomer as the major component,some cyclization product was expected. The iodide 11 was recovered upon treatment with milder bases,while decomposed products were detected upon treatment with harsher bases. The reluctance of this ring closure may be due to the strain energy associated with 1,3- diaxial repulsion between the methyl group and the forming C1- C2 bond of the transition state 12 (Scheme 5).

|
Download:
|
| Scheme 5. Preliminary efforts toward the synthesis of the tricyclic framework. | |
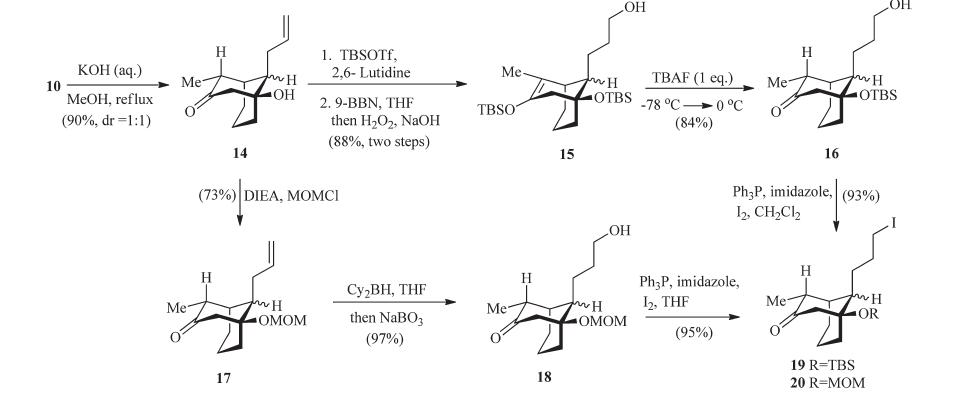
Bearing in mind the severe 1,3-diaxial repulsion,it was considered a wise decision to remove the extra methyl ester group in an earlier stage in the modified synthesis (Scheme 6). Treatment of a 1:4.3 mixture of 10 with refluxing methanolic KOH (3 equiv.,24 h) produced the decarboxylation product 14 as an equal amount of diastereomeric mixture. We propose that this isomerization takes place via a retro-aldol reaction followed by base catalyzed epimerization of the ring-opened diketones [19]. Compound 14 was then treated with excess TBSOTf (2.2 equiv.),the doubly silyl ether protected productwas formed as the sole product in good yield. It is worth noting that the carbonyl group of 14 is as reactive as the hydroxyl group. The doubly protected compound was isolated as the major product (~40% yield) when little excess amount of TBSOTf (1.2 equiv.) was utilized. Next,chemoselective hydroboration of the allyl double bond followed by oxidation afforded the alcohol 15. Chemoselective removal of the enol ether TBS group offered the primary alcohol 16.

|
Download:
|
| Scheme 6. Synthesis of the precursor. | |
In an effort to explore the role of protective groups in the synthesis of the tricyclic compound,MOM ether 17 was obtained by mixing the ketone alcohol 14 with MOMCl in the presence of DIEA (Scheme 6). Next,chemoselective hydroboration-oxidation of the allyl double bond was achieved to furnish the primary alcohol 18 in 97% yield by treatingMOMether 17 with dicyclohexylborane followed by oxidative workup [21]. It is worth noting that the borane reagent is crucial to achieve excellent selectivity. Both 9- BBN and BH3 react preferentially with the carbonyl group of 17. Iodinization of the alcohol 16 or 18 afforded the iodide 19 or 20 respectively.
With the iodide in hand,efforts were focused on the final ring closure reaction. This tricky cyclization reaction has been carefully investigated. In order to obtain the desired tricyclic product 21 or 23,we tried LDA,a kinetically fast base at low temperature. Surprisingly,no reaction was detected when iodide 19 was treated with LDA at -78 °C (Table 1,entry 1). Gratefully, the desired tricyclic compound 21 was formed as the major product (ratio = 2.3:1) when the reaction temperature was increased to -40 °C (entry 2). Furthermore,we were able to isolate the two regioisomeric products 21,22 and the unreactive starting material 19b. As expected,the ratio dropped as the temperature increased. At 0 °C,both the ratio and the yield were reduced (entry 3). The temperature/base pair of -15 °C/LiTMP appears to be optimal (entry 5). This ring closure reaction forMOM ether 20 showed poorer selectivity in comparison with that for TBS ether 19 (entry 7).
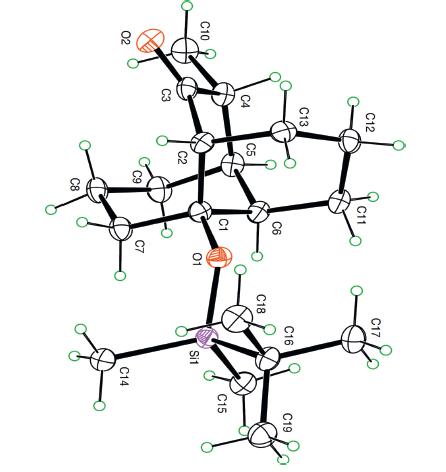
The structure and the relative stereochemistry of 21 have been verified by X-ray crystallographic analysis (Fig. 1) [22].
It is worth noting that no reaction occurred when 19 was treated with LiTMP at -40 °C (Table 1,entry 4),whereas the reaction was finished in one hour at the same temperature utilizing LDA. This result indicates that the formation of enolate rather than the substitution of iodide is the rate determining step.

|
Download:
|
| Fig. 1. ORTEP plot of the final product 21. | |
| Table 1 Regioisomer distribution in the cyclization reaction. |

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
In summary,two compounds with tricyclo[6.4.0.04,9]dodecane framework has been successfully constructed in seven or eight linear steps. An innovative strategy accessing the framework from bicyclo[3.3.1]nonanes was employed. More than one gram of the tricyclic compound has been obtained in 13% overall yield,which allows the synthesis of the unusual skeleton to be a practical one. The approach employs a Robinson annulation,a base induced decarboxylation and epimerization in a single step,and an intramolecular alkylation that allows removal of the unreactive isomer from the Robinson annulation. This synthesis not only provides an access to the tricyclo[6.4.0.04,9]dodecane framework, but also demonstrates the utility of our previous investigations in one-carbon bridge stereocontrol in Robinson annulations leading to bicyclo[3.3.1]nonanes. Furthermore,the efficient approach could also be a potential access to other types of cis-decalin bearing a 1,5-bridging framework.
AcknowledgmentWe thank Frank Fronczek (LSU) for assistance with X-ray crystallography. Financial support from TianJin Municipal Science and Technology Commission (No. 11ZCKFSY06200) is greatly acknowledged.
| [1] | I. Uchida, T. Ando, N. Fukami, et al., The structure of vinigrol, a novel diterpenoid with antihypertensive and platelet aggregation-inhibitory activities, J. Org. Chem. 52 (1987) 5292-5293. |
| [2] | T. Ando, Y. Tsurumi, N. Ohata, et al., Vinigrol, a novel antihypertensive and platelet aggregation inhibitory agent produced by a fungus, Virgaria nigra. I. Taxonomy, fermentation, isolation, physicochemical and biological properties, J. Antibiot. 41 (1988) 25-30. |
| [3] | T. Ando, K. Yoshida, M. Okuhara, Vinigrol, a novel antihypertensive and platelet aggregation inhibitory agent produced by a fungus, Virgaria nigra. II. Pharmacological characteristics, J. Antibiot. 41 (1988) 31-35. |
| [4] | H.G. Fritz, H. Henke, H. Musso, Asteranes. XI. 1,4-Dihydroxytricyclo[6.4.0.04,9]-dodecane-7,10-dione from furanophane, Chem. Ber. 107 (1974) 3164-3175. |
| [5] | H. Buding, H. Musso, Optically active tricyclo[6.4.0.04,9]dodecane, Angew. Chem. Int. Ed. 17 (1978) 851. |
| [6] | H. Buding, B. Deppisch, H. Musso, G. Snatzke, (R)-and (S)-tricyclo[6.4.0.04,9]-dodecane, Chem. Ber. 118 (1985) 4597-4612. |
| [7] | H. Buding, B. Fuchs, H. Musso, Elimination of halogen from 1,4-dibromotricyclo[ 6.4.0.04,9]dodecane, Chem. Ber. 118 (1985) 4613-4615. |
| [8] | E. Osawa, K. Aigami, Y. Inamoto, Application of force field calculations to organic chemistry. 6. Steric analysis of synthesis and structure of 1,4-dihydroxytricyclo[ 6.4.0.04,9]dodecane-7,10-dione. Dynamic conformational calculations of its hydrocarbon skeleton and related systems (bicyclo[3.3.1]nonane and bicyclo[ 3.3.2]decane), J. Chem. Soc., Perkin Trans. 2 (1979) 172-180. |
| [9] | H.E. Winberg, F.S. Fawcett, W.E. Mochel, C.W. Theobald, Dimethylenedihydroheteroaromatic compounds and heterocyclophanes by 1, 6-Hofmann elimination reaction, J. Am. Chem. Soc. 82 (1960) 1428-1435. |
| [10] | Y. Ito, S. Miyata, M. Nakatsuka, T. Saegusa, Fluoride-induced 1,6-elimination to pquinodimethane. A new preparative method for [2.2]paracyclophane,[2.2](2.5)furanophane and [2.2](2.5)thiophenophane, J. Org. Chem. 46 (1981) 1043-1044. |
| [11] | P.S. Chen, C.H. Chou, Synthesis and chemistry of 2,5-dimethylene-2,5-dihydrofuran, J. Chin. Chem. Soc. (Taipei) 39 (1992) 251-255. |
| [12] | K. Vandyck, B. Matthys, J. Van der Eycken, Synthesis and absolute configuration of (1S, 8S)-as-hydrindacene-1,8-diol as determined by the circular dichroism exciton chirality method, Tetrahedron Lett. 46 (2005) 75-78. |
| [13] | L.A. Paquette, R. Guevel, S. Sakamoto, I.H. Kim, J. Crawford, Convergent enantioselective synthesis of vinigrol, an architecturally novel diterpenoid with potent platelet aggregation inhibitory and antihypertensive properties. 1. Application of anionic sigmatropy to construction of the octalin substructure, J. Org. Chem. 68 (2003) 6096-6107. |
| [14] | L.A. Paquette, I. Efremov, Z. Liu, Exploratory studies aimed at a synthesis of vinigrol. 2. Attempts to exploit ring-closing metathesis for construction of the central cyclooctane belt, J. Org. Chem. 70 (2005) 505-509. |
| [15] | L.A. Paquette, I. Efremov, Exploratory studies aimed at a synthesis of vinigrol. 3. Evaluation of a lactone bridge as a conformational lock, J. Org. Chem. 70 (2005) 510-513. |
| [16] | L.A. Paquette, Z. Liu, I. Efremov, Exploratory studies aimed at a synthesis of vinigrol. 4. Probe of possible means for direct connection of the side arms and of ring-contraction alternatives, J. Org. Chem. 70 (2005) 514-518. |
| [17] | J.E. Baldwin, Rules for ring closure, J. Chem. Soc. Chem. Commun. (1976) 734-736. |
| [18] | J.E. Baldwin, M.J. Lusch, Rules for ring closure: application to intramolecular aldol condensations in polyketonic substrates, Tetrahedron 38 (1982) 2939-2947. |
| [19] | D. Wang, W.E. Crowe, One-carbon bridge stereocontrol in Robinson annulations leading to bicyclo[3.3.1]nonanes, Org. Lett. 12 (2010) 1232-1235. |
| [20] | X. Jiang, X. Xu, F. Qing, Design and concise synthesis of gem difluoromethylenated analogue of 7-epi-castanospermine, Chin. Chem. Lett. 25 (2014) 1115-1120. |
| [21] | G.W. Kabalka, S. Yu, N.S. Li, Selective hydroboration of terminal alkenes in the presence of aldehydes and ketones, Tetrahedron Lett. 38 (1997) 5455-5458. |
| [22] | CCDC-1012577 contains supplementary crystallographic data for this paper. These data can be obtained free of charge from The Cambridge Crystallographic Data Centre via www.ccdc.cam.ac.uk/data_request/cif. |