




b Division of Anti-tumor Pharmacology, State Key Laboratory of Drug Research, Shanghai Institute of Materia Medica, Chinese Academy of Sciences, Shanghai 201203, China;
c Department of Medicinal Chemistry, Shanghai Institute of Materia Medica, Chinese Academy of Sciences, Shanghai 201203, China
Phosphatidylinositol-3-kinases (PI3Ks) [1] play an important role in signal transduction as a key regulator of cell cycle proliferation,growth,survival and protein synthesis [2],and they catalyze phosphorylation of 3-hydroxyl position of phosphatidylinositides (PIs) [3, 4, 5]. Based on their primary structure and mechanism of action [6],PI3Ks are divided into three major classes [7]: class I,II,and III [8]. Class I is further split into class Ia: p110a, p110β,and p110δ,which are activated by tyrosine kinase receptors; and class Ib: p110g,which is activated by G proteincoupled receptor [9]. The PIK3CA gene that encodes p110a is also frequently mutated in many cancers. Thus class Ia PI3Ks,and particularly p110a,are potential therapeutic targets for cancers [10].
Wortmannin [11] and LY-294002 [12] have been extensively studied,but both of them lack selectivity over the class I PI3K isoforms (Fig. 1). In recent years,many small molecular inhibitors have entered into preclinical or clinical stage [13],such as compound 1 (PIK-75) [7] and 2 (PKI-587) [14]. Compound 1 (PIK-75) was reported to be a sub-nanomolar p110a inhibitor with more than100-fold selectivity over p110b and p110g,and it also showed activity in human cancer xenograft model.

|
Download:
|
| Fig. 1. Some reported PI3K inhibitors. | |
The work we reported here investigated the structure-activity relationship (SAR) of compound 1 (PIK-75) by exploring major changes on the bicyclic core structure and the side chains. We intended to determine whether the position of substituents or the number of the nitrogen atom on the ring was crucial,and to test the influence of the variants in side chain. In this regard,a series of acylhydrazone analogs were synthesized and tested in enzymatic and cellular levels. 2. Experimental
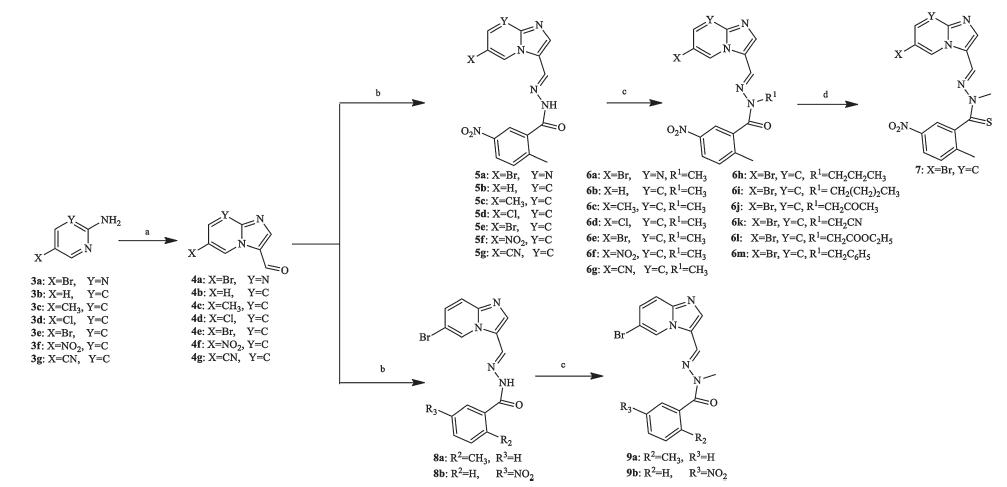
The synthesis of compounds 6a-m,7,and 9a-b was shown in Scheme 1. Aldehydes 4a-g were prepared from 3a-g by cyclization with bromomalonaldehyde. Compounds 6a-m and 9a-b were synthesized by condensation of the aldehydes with substituted benzohydrazide followed by alkylation. Compound 7 was obtained from 6e by replacement of oxygen with sulfur using Lawesson’s reagent. The synthesis of compound 12 was shownin Scheme 2. The structure of the new analogs was characterized by 1H NMR and MS. In addition,the structure of the analogs (represented by compound 6e) was also confirmed by single crystal X-ray diffraction.

|
Download:
|
| Scheme 1. Synthesis of compounds 6a-m,7,and 9a-b. Reagents and conditions: (a) bromomalonaldehyde,EtOH,reflux,12 h; (b) benzohydrazide,HOAc (cat.),MeOH,60 °C, 3 h; (c) NaH,R-X,THF-DMF,r.t.,1 h; (d) Lawesson’s reagent,dioxane,reflux,12 h. | |

|
Download:
|
| Scheme 2. Synthesis of compound 12. Reagents and conditions: (a) benzohydrazide,HOAc (cat.),MeOH,60 °C,3 h; (b) NaH,MeI,THF,r.t.,1 h. | |
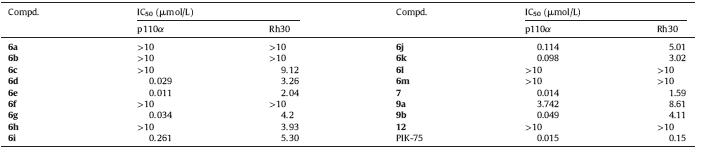
We firstly investigated the influence of the position of substituents and number of nitrogen atoms on the ring in the compounds on their activity. Compounds 6a and 12 showed poor activity against p110α (IC50 > 10 μmol/L) and Rh30 (IC50 > 10 mmmol/ L) (Table 1). Compound 6e displayed good activity with an IC50 of 11 nmol/L (Table 2). The results indicated that the position of substituents and the number of nitrogen atoms on the bicyclic core structure were crucial to maintain both enzymatic and cellular activities for this series of compounds.
| Table 1 Biological activity of compounds 6a-m,7,9a-b and 12. |
{kind=link}
{kind=link}
{kind=link}
| Table 2 Kinase-selectivity profiling of compound 7.a |
Next,various groups at the C6 position of the imidazo[1,2- α]pyridine moiety were screened (Table 1). Compound 6b,without any substituent at C6 position,showed poor activity. Introduction of a methyl group as electron donor led to reduced potency. The less bulky chloro-substituted derivative (6d) and the derivatives with strong electron-withdrawing groups NO2 and CN (6f and 6g) were less potent than 6e. We envisioned that the bromine atom played a critical role in compound binding with PI3K due to the reason that compound with a bromine substitution may fit well in the active site of PI3K.
With an optimized bromo substitution on bicyclic ring,various R1 groups were screened. Compounds 6h and 6i with longer carbon chain were less potent than 6e. Compounds with bulky substitution (6l and 6m) had very poor potency (IC50 > 10 mmol/L). The results indicated that the length and the size of the substituents had a substantial influence on activity. It was to our delight that compound 7,with sulfur replacement of carbonyl oxygen,showed good activity against p110α and Rh30 (IC50: 14 nmol/L and 1.59 mmol/L,respectively). Removal of the nitro group on benzene ring (9a) led to a sharp drop in p110a activity (IC50: 3.75 mmol/L) compared to 9b (IC50: 49 nmol/L).
Finally,compound 7,the representative compound in this series of compounds,was further evaluated on a panel of tyrosine kinases (Table 2),and it was inactive against other kinases, indicating that it was a selective PI3K inhibitor. 4. Conclusion
In summary,a series of acylhydrazone derivatives were synthesized,and they were identified as potential PI3K inhibitors with no apparent inhibition on a panel of other kinases. Therefore, our results indicated that this class of compounds could be served as lead compound for development of more selective anticancer medication.
AcknowledgmentsWe thank the National Natural Science Foundation of China (Nos. 81273365 and 81321092), Chinese National Science & Technology Major Project ‘‘Key New Drug Creation and Manufacturing Program’’ (Nos. 2012ZX09103101-024 and 2014ZX09304002-008-001), Chinese National Programs for High Technology Research and Development (No. 2012AA020302) and the Shanghai Science and Technology Commission (Nos. 11431921100 and 12DZ1930802) for their financial support.
| [1] | T.A. Yap, M.D. Garret, M.I. Walton, et al., Targeting the PI3K-AKT-mTOR pathway: progress, pitfalls, and promises, Curr. Opin. Pharmacol. 8 (2008) 393-412. |
| [2] | I. Vivanco, C.L. Sawyers, The phosphatidylinositol 3-kinase AKT pathway in human cancer, Nat. Rev. Cancer 2 (2002) 489-501. |
| [3] | M. Hayakawa, H. Kaizawa, K. Kawaguchi, et al., Synthesis and biological evaluation of imidazo[1,2-a]pyridine derivatives as novel PI3 kinase p110a inhibitors, Bioorg. Med. Chem. 15 (2007) 403-412. |
| [4] | J.D. Kendall, A.C. Giddens, K.Y. Tsang, et al., Novel pyrazolo[1,5-a]pyridines as p110α-selective PI3 kinase inhibitors: exploring the benzenesulfonohydrazide SAR, Bioorg. Med. Chem. 20 (2012) 58-68. |
| [5] | L.C. Cantley, The phosphoinositide 3-kinase pathway, Science 296 (2002) 1655-1657. |
| [6] | M. Hayakawa, K. Kawaguchi, H. Kaizawa, et al., Synthesis and biological evaluation of sulfonylhydrazone-substituted imidazo[1,2-a]pyridines as novel PI3 kinase p110a inhibitors, Bioorg. Med. Chem. 15 (2007) 5837-5844. |
| [7] | B. Vanhaesebroeck, M.D. Waterfield, Signaling by distinct classes of phosphoinositide 3-kinases, Exp. Cell Res. 253 (1999) 239-254. |
| [8] | M.A. Lawlor, D.R. Alessi, PKB/Akt: a key mediator of cell proliferation, survival and insulin responses? J. Cell Sci. 114 (2001) 2903-2910. |
| [9] | D.A. Fruman, R.E. Meyers, L.C. Cantley, Phosphoinositide kinases, Annu. Rev. Biochem. 67 (1998) 481-507. |
| [10] | Y. Samuels, Z. Wang, A. Bardelli, et al., High frequency of mutations of the PIK3CA gene in human cancers, Science 304 (2004) 554. |
| [11] | B.H. Norman, C. Shih, J.E. Toth, et al., Studies on the mechanism of phosphatidylinositol 3-kinase inhibition by wortmannin and related analogs, J. Med. Chem. 39 (1996) 1106-1111. |
| [12] | C.J. Vlahos, W.F. Matter, K.Y. Hui, et al., A specific inhibitor of phosphatidylinositol 3-kinase, 2-(4-morpholinyl)-8-phenyl-4H-1-benzopyran-4-one (LY294002), Biol. Chem. 269 (1994) 5241-5248. |
| [13] | W.F. Zhu, X. Zhai, S. Li, et al., Synthesis and cytotoxic activity of novel 2,6- disubstituted-4-mor-pholinothieno[3,2-d]pyrimidines as potent anti-tumor agents, Chin. Chem. Lett. 23 (2012) 703-706. |
| [14] | A.M. Venkatesan, C.M. Dehnhardt, E.D. Santos, et al., Bis(morpholino-1,3,5-triazine) derivatives: potent adenosine 5'-triphosphate competitive phosphatidylinositol- 3-kinase/mammalian target of rapamycin inhibitors: discovery of compound 26 (PKI-587), a highly efficacious dual inhibitor, J. Med. Chem. 53 (2010) 2636-2645. |