




b Department of Medicinal Chemistry, The University of Kansas, Lawrence KS66045, USA
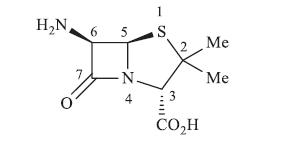
Penicillins are β-lactam antibiotics to kill many gram-positive, gram-negative and anaerobic organisms by blocking peptidoglycan biosynthesis [1, 2, 3, 4, 5, 6, 7, 8]. β-Lactams are 4-memebered cyclic amides, depicted in 6-aminopenicillanic acid (6-APA) in Fig. 1 [9, 10]. Various bonds in β-lactam can undergo cleavage to give acyclic systems or result into rearranged cyclic derivatives [11, 12, 13, 14, 15]. Cleavage of β-lactam bond (N4-C7) and conversion of β-lactam ring into other cyclic systems have been studied [16, 17, 18]. Nucleophilic opening of β-lactam bond using a primary amine forms thiazolidine amides [19, 20].

|
Download:
|
| Fig. 1. Chemical structure of 6-aminopenicillanic acid (6-APA). | |
{kind=link}
In this paper,chemical modification of penicillin β-lactam ring was undertaken. β-Lactam ring rearrangement of 6-APA produced 8-hydroxypenillic acid derivatives with side chains of methyl, propyl,benzyl,diethylaminoethyl,and 2-(bromomethyl)benzo[ d]thiazole groups. β-Lactam ring opening of penicillin V methyl ester yielded thiazolidine amides with p-methoxybenzylamine, anisidine,benzyl amine,thiophene methylamine,cyclopropane methyl amine,and nonyl amine. Parallel synthetic methods were developed for esterification of 8-hydroxypenillic acid and β-lactam ring opening of penicillin V methyl ester. 2. Experimental
Column chromatography was carried out by employing silica gel (230-400 mesh). Thin-layer chromatography (TLC) was performed on a silica gel w/uv254 uniplateTM. Anhydrous organic solvents were purchased. Parallel synthesis was conducted on Mettler Toledo MiniBlock and MiniBlock XT. Melting points were determined using a Barnstead International MET-TEMP1 capillary melting point apparatus model 1001D-120VAC. IR spectra were measured with a Perkin ElmerTM Spectrum One FT-IR spectrometer. 1H NMR and 13C NMR spectra were recorded on a 400 MHz spectrometer (400 and 100 MHz,respectively),or a 500 MHz spectrometer (500 and 125.5 MHz,respectively). Abbreviations were as follows: s,singlet; d,doublet; t,triplet; q,quartet; m, multiplet. High-resolution mass spectrometry (HRMS) spectra were obtained on a double-focusing mass spectrometer. 2.1. Procedure for synthesis of intermediate penicillin V
Potassium (2S,5R,6R)-3,3-dimethyl-7-oxo-6-(2-phenoxyacetamido)- 4-thia-1-azabicyclo[3.2.0]heptane-2-carboxylate (penicillin V). To a cooled and stirred solution of 2.76 g (12.5 mmol) of 6-APA in 60 mL of water containing 5.25 g (62.5 mmol) of sodium bicarbonate,a solution of 2.76 g (16.2 mmol) phenoxyacetyl chloride in 5 mL of acetone was added in one minute. The resulting mixture was stirred vigorously during 20 min while the temperature was kept at 10-15 °C. The clear solution was extracted twice with 15 mL portions of methyl isobutyl ketone (MIBK). The clear aqueous solution was cooled to 5-10 °C and acidified to pH 2 with a cold 5.0 mol/L sulfuric acid solution. The acidified aqueous solution was extracted with 50 mL MIBK twice. The MIBK extract was separated,washed with cold water,and dried for 10 min over anhydrous sodium sulfate. After filtration, 10 mL of a 25% solution of potassium 2-ethylhexanoate in butanol was added. The white crystalline material was collected by filtration,washed on the filter with dry acetone and dried in vacuum,yield 3.5 g (80%) penicillin V as a white solid. Mp: 210- 211 °C (dec.). 1H NMR (400 MHz,DMSO-β6): δ 8.42 (d,1H, J = 8.0 Hz),7.27 (m,2H),6.92 (m,3H),5.42 (dd,1H,J = 8.0 Hz, 4.0 Hz),5.40 (d,1H,J = 4.0 Hz),4.62 (d,2H,J = 2.2 Hz),3.88 (s,1H), 1.52 (s,3H),1.46 (s,3H); 13C NMR (100 MHz,DMSO-β6): δ 172.8, 169.0,168.1,158.1,130.0,121.7,115.0,74.6,67.3,66.7,65.0,57.8, 32.7,27.8; HRMS (FAB) calcd. for C16H18KN2O5S [M+H]+: m/z 389.0584; found: 389.0995. 2.2. Procedure for synthesis of intermediate penicillin V methyl ester (1)
(2S,5R,6R)-Methyl 3,3-dimethyl-7-oxo-6-(2-phenoxyacetamido)- 4-thia-1-azabicyclo[3.2.0]heptane-2-carboxylate (1): Penicillin V (388 mg,1.0 mmol) was suspended in 10 mL of dimethylformamide (DMF). Methyl iodide (1.4 g,12.0 mmol) was added and stirred for 1 h at room temperature. After most of the solvent was removed under reduced pressure,the mixture was loaded on silica gel column for chromatography with EtOAc/hexane (1:9) as an eluent to yield product 1 (65%) as a colorless oil. 1HNMR (400 MHz, CDCl3): δ 7.32 (m,2H),7.04 (m,1H),6.93 (m,2H),5.75 (d,1H, J = 4.0 Hz),5.60 (d,1H,J = 4.0 Hz),4.57 (s,2H),4.49 (s,1H),3.80 (s, 3H),1.61 (s,3H),1.51 (s,3H); 13C NMR (100 MHz,CDCl3): δ 173.1, 168.1,167.8,156.9,129.8,122.4,114.8,70.5,67.7,67.1,64.8,58.0, 52.5,31.8,26.9; HRMS (FAB) calcd. for C17H21N2O5S [M+H]+: m/z 365.1170; found: 365.1194. 2.3. Procedure for synthesis of thiazolidine derivatives 2a-2f
(2R,4S)-Methyl 2-((R)-2-(4-methoxybenzylamino)-2-oxo-1-(2- phenoxyacetamido)ethyl)-5,5-dimethylthiazolidine-4-carboxylate (2a): Penicillin V methyl ester (1,182.0 mg,0.5 mmol) was taken in a round-bottomed flask and 15 mL of dry methylene chloride was added. Benzyl amine (108.0 mg,1.0 mmol) was added at room temperature and the mixture was stirred overnight. Water was added into the mixture and the mixture was extracted with methylene chloride (2 × 20 mL). After washing with brine,the organic layer was dried over NaSO4,concentrated and purified by column chromatography with EtOAc/hexane (1:1) as the eluent to yield product 2a (55%) as a semi-solid. 1HNMR (400 MHz,CDCl3): d 7.59 (d,1H,J = 7.36 Hz),7.33-7.29 (m,5H),7.01 (m,2H),6.93 (m, 2H),5.27 (s,1H),4.65 (m,1H),4.61 (m,1H),4.51 (s,2H),4.37 (dd, 1H,J = 5.9,14.7 Hz),3.75 (s,3H),3.55 (s,2H),1.48 (s,3H),1.20 (s, 3H); 13CNMR(100 MHz,CDCl3): δ 169.9,168.9,168.5,157.1,137.7, 129.7,128.8,127.9,127.6,122.1,114.8,72.6,67.2,65.4,58.0,56.8, 52.3,43.8,26.6,26.4; HRMS (FAB) calcd. for C24H30N3O5S [M+H]+: m/z 472.1906; found: 472.1910.
(2R,4S)-Methyl 2-((R)-2-(4-methoxyphenylamino)-2-oxo-1- (2-phenoxyacetamido)ethyl)-5,5-dimethylthiazolidine-4-carboxylate (2b): Yield 62%; 1H NMR (400 MHz,CDCl3): δ 8.59 (s,1H), 7.67 (d,1H,J = 6.4 Hz),7.42 (m,2H),7.32 (m,2H),6.96 (m,1H), 6.95 (m,2H),6.85 (m,2H),5.36 (m,1H),4.74 (m,1H),4.57 (s,2H), 3.80 (s,3H),3.75 (s,3H),3.64 (s,1H),1.55 (s,3H),1.24 (s,3H); 13C NMR (100 MHz,CDCl3): δ 169.8,169.3,166.6,157.1,156.7,129.5, 129.8,122.2,121.8,114.8,114.2,72.8,67.3,65.3,58.0,57.3,55.5, 52.4,26.8,26.5; HRMS (FAB) calcd. for C24H30N3O6S [M+H]+: m/z 488.1855; found: 488.1849.
(2R,4S)-Methyl 5,5-dimethyl-2-((R)-2-oxo-1-(2-phenoxyacetamido)- 2-(thiophen-3-ylmethylamino)ethyl)thiazolidine-4-carboxylate (2c): Yield: 63%; 1H NMR (400 MHz,CDCl3): δ 7.61 (d,1H, J = 7.56 Hz),7.32 (m,3H),7.18 (s,1H),7.01 (m,2H),6.91 (m,3H), 5.24 (d,1H,J = 4.9 Hz),4.72 (m,1H),4.62 (m,1H),4.52 (m,1H),4.50 (m,2H),3.73 (s,3H),3.56 (s,2H),1.47 (s,3H),1.18 (s,3H); 13C NMR (100 MHz,CDCl3): δ 169.8,168.9,168.4,157.1,140.2,129.7,126.9, 126.2,125.2,122.1,114.8,72.5,67.2,65.5,58.0,56.7,52.2,38.2, 26.6,26.5; HRMS (FAB) calcd. for C22H28N3O5S2 [M+H]+: m/z 478.1470; found: 478.1475.
(2R,4S)-Methyl 2-((R)-2-(benzylamino)-2-oxo-1-(2-phenoxyacetamido) ethyl)-5,5-dimethylthiazolidine-4-carboxylate (2d): Yield 54%; 1H NMR (400 MHz,CDCl3): δ 7.48 (d,1H,J = 7.4 Hz), 7.46 (d,1H,J = 8.0 Hz),7.21 (m,2H),7.20 (m,2H),7.03 (m,1H),6.99 (m,2H),6.86 (m,2H),5.36 (m,1H),5.13 (m,1H),4.68 (m,1H),4.64 (m,1H),4.57 (m,2H),4.23 (m,1H),3.77 (s,3H),3.73 (s,3H),1.48 (s, 3H),1.18 (s,3H); 13C NMR (100 MHz,CDCl3): δ 172.8,169.8,168.3, 156.8,137.8,129.8,128.6,127.4,122.0,115.6,74.9,73.0,72.4,65.1, 58.0,56.8,52.3,43.6,26.5,18.9; HRMS (FAB) calcd. for C25H32N3O6S [M+H]+: m/z 5 02.2011; found: 502.1992.
(2R,4S)-Methyl 2-((R)-2-(cyclopropylamino)-2-oxo-1-(2-phenoxyacetamido) ethyl)-5,5-dimethylthiazolidine-4-carboxylate (2e): Yield 64%; 1H NMR (400 MHz,CDCl3): δ 7.55 (d,1H, J = 7.7 Hz),7.29 (m,2H),7.01 (m,2H),6.80 (m,1H),5.17 (d,1H, J = 8.0 Hz),4.51 (s,1H),4.50 (s,2H),3.75 (s,3H),3.66 (s,1H),2.70 (m,1H),1.53 (s,3H),1.19 (s,3H),0.73 (m,2H),0.49 (m,2H); 13C NMR (100 MHz,CDCl3): δ 170.1,169.8,168.9,157.2,129.7,122.1, 114.8,72.7,67.2,65.8,58.3,56.8,52.3,27.0,26.6,22.7,6.5,6.4; HRMS (FAB) calcd. for C20H28N3O5S [M+H]+: m/z 422.1749; found: 422.1739.
(2R,4S)-Methyl 5,5-dimethyl-2-((R)-2-(nonylamino)-2-oxo-1- (2-phenoxyacetamido)ethyl)thiazolidine-4-carboxylate (2f): Yield 58%; 1H NMR (400 MHz,CDCl3): δ 7.58 (d,1H,J = 8 Hz), 7.24 (m,2H),6.98 (m,1H),6.90 (m,2H),6.84 (m,1H),5.15 (m, 1H),4.57 (m,1H),4.48 (s,1H),3.71 (s,3H),3.68 (s,1H),3.51 (m, 1H),3.17 (m,2H),1.51 (s,3H),1.45 (m,2H),1.21 (m,14H),1.17 (s, 3H),0.83 (m,3H); 13C NMR (100 MHz,CDCl3): δ 169.8,168.8, 168.7,157.2,129.6,122.0,114.8,72.6,67.3,66.0,58.3,57.0,52.1, 39.7,31.8,29.5,29.4,29.3,29.2,27.1,26.9,26.7,22.6,14.1; HRMS (FAB) calcd. for C26H42N3O5S [M+H]+ : m/z 508.2845; found: 508.2828. 2.4. Procedure for synthesis of intermediate disodium salt of 8-hydroxypenillic acid (3)
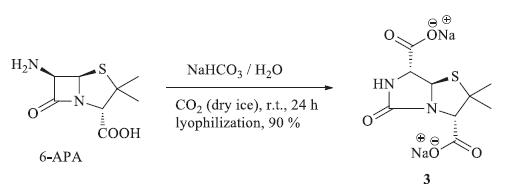
Disodium 3,3-dimethyl-8-oxo-4-thia-1,7-diazabicyclo[3.3.0]- octane-2,6-dicarboxylic acid (3): Compound 3 was prepared in the method modified from Johnson and Hardcastle [17]. 4.5 g of 6- APA was dissolved in 100 mL of water containing 3.5 g (2.0 equiv.) of sodium bicarbonate. Carbon dioxide from dry ice was bubbled through the stirred mixture at room temperature for 24 h. The concentrated aqueous solution was then lyophilized overnight to yield 6.2 g (90%) of the product 3 as a pale yellow powder. Mp 228- 230 °C (dec.). 1H NMR (400 MHz,D2O): δ 5.44 (d,1H,J = 2.0 Hz), 4.15 (s,1H),4.13 (d,1H,J = 2.0 Hz),1.47 (s,3H),1.42 (s,3H); 13C NMR (125 MHz,D2O): δ 177.5,175.9,164.4,73.6,69.4,59.5,57.7, 31.5,25.8; HRMS (FAB) calcd. for C9H11N2Na2O5S [M+H]+: m/z 305.0184; found: 305.0174. 2.5. Procedure for alkylation of 8-hydroxypenillic acid to synthesize compounds 4a-4d
(3S,7R,7aR)-Dimethyl 2,2-dimethyl-5-oxohexahydroimidazo [5, 1, b]thiazole-3,7-dicarboxylate (4a): Compound 4a was prepared in the method of Johnson and Hardcastle [17]. The synthesis of the dimethyl ester was initiated by dissolving the disodium salt 3 (2.0 g,6.6 mmol) in DMF (50 mL) and methyl iodide (3.0 mL,48.1 mmol) was added and stirred at room temperature for 12 h,water was added to the reaction mixture and extracted with (3 × 100 mL) diethyl ether,dried and concentrated. The crude product was purified by flash silica gel column chromatography using EtOAc/hexane (1:1) as an eluent. Pure compound 4a (1.14 g,60%)was isolated as awhite solid.Mp: 164-166 °C. 1H NMR (400 MHz,CDCl3): δ 5.89 (s,1H),5.78 (d,1H, J = 1.48 Hz),4.70 (s,1H),4.35 (d,1H,J = 1.52 Hz),3.85 (s,3H),3.78 (s,3H),1.57 (s,3H),1.49 (s,3H); 13C NMR (100 MHz,CDCl3): d 170.2,169.3,161.2,70.8,68.2,58.6,58.3,53.2,52.1,33.7, 26.4. HRMS (FAB) calcd. for C11H17N2O5S [M+H]+: m/z 289.0788; found: 289.0864.
(3S,7R,7aR)-Dipropyl 2,2-dimethyl-5-oxohexahydroimidazo [5, 1, b]thiazole-3,7-dicarboxylate (4b): Yield 62%; mp 119- 120 °C; 1H NMR (400MHz,CDCl3): δ 6.20 (s,1H),5.78 (s,1H), 4.69 (s,1H),4.33 (s,1H),4.16 (d,2H,J = 6.72 Hz),4.10 (dd,2H, J = 6.8,13.5 Hz),1.66 (m,4H),1.55 (s,3H),1.48 (s,3H),0.95 (q,6H, J = 8.0 Hz); 13CNMR (100MHz,CDCl3): δ 169.9,168.9,161.5,70.8, 68.4,67.9,66.9,58.6,58.5,34.0,26.4,21.8,10.5,10.2; HRMS (FAB) calcd. for C15H25N2O5S [M+H]+: m/z 345.1484; found: 345.1496.
(3S,7R,7aR)-Dibenzyl 2,2-dimethyl-5-oxohexahydroimidazo [5, 1, b]thiazole-3,7-dicarboxylate (4c): Yield 63%; mp 133- 134 °C; 1H NMR (400 MHz,CDCl3): δ 7.35-7.28 (m,10H),5.95 (s,1H),5.83 (d,1H,J = 1.4 Hz),5.22 (m,2H),5.17 (s,2H),4.74 (s, 1H),3.37 (d,1H,J = 1.36 Hz),1.54 (s,3H),1.42 (s,3H); 13C NMR (100 MHz,CDCl3): δ 169.6,168.7,161.3,134.9,134.7,128.8, 128.7,128.6,128.5,70.7,68.3,68.0,67.2,58.8,58.4,33.9,26.4; HRMS(FAB) calcd. for C23H25N2O5S [M+H]+ : m/z 441.1484; found: 441.1485.
(3S,7R,7aR)-Bis(2-(diethylamino)ethyl) 2,2-dimethyl-5-oxohexahydroimidazo[ 5,1-b]thiazole-3,7-dicarboxylate (4d): To a suspension of N,N-diethylaminoethyl bromide hydrobromide (1.05 g,4.0 mmol) in 15 mL of dry DMF was added sodium bicarbonate (1.26 g,15 mmol). The suspension was stirred at room temperature for 2 h,then disodium salt of 8-hydroxypenillic acid 3 (500 mg,1.6 mmol) was added. The reaction mixture was allowed to stir at room temperature overnight. Water (10 mL) was added to the reaction mixture and extracted with diethyl ether (3 × 50 mL). The organic layer was dried over Na2SO4 and concentrated to obtain a colorless semi-solid 4d 189 mg (40%). IR: 3236,3104, 2972,2807,1731,1613,1456,1382,1176,1122 cm-1. 1H NMR (400 MHz,CDCl3): δ 6.65 (s,1H),5.73 (d,1H,J = 1.58 Hz),4.64 (s, 1H),4.32(d,1H,J = 1.56 Hz),4.24 (t,2H,J = 5.64 Hz),4.17 (t,2H, J = 6.0 Hz),2.74-2.67 (m,4H),2.60-2.51 (m,8H),1.53 (s,3H),1.47 (s,3H),1.02-0.97 (m,12H); 13C NMR (100 MHz,CDCl3): δ 169.9, 168.8,161.5,70.8,68.3,64.0,58.3,58.5,51.0,50.8,47.3,47.2,33.7, 26.4,11.9,11.6; HRMS (FAB) calcd. for C21H39N4O5S [M+H]+: m/z 459.2641; found: 459.2640. 2.6. Procedure for alkylation of 8-hydroxypenillic acid methyl ester at position N-7 to synthesize compounds 5a and 5b
(3S,7R,7aR)-Dimethyl 6-benzyl-2,2-dimethyl-5-oxohexahydroimidazo[ 5,1-b]thiazole-3,7-dicarboxylate (5a): Benzyl bromide (2.05 g,1.2 mmol) and anhydrous K2CO3 (2.5 mg,1.8 mmol) were added to a vigorously stirred solution of 4a (200 mg,0.7 mmol) in DMF and stirring was continued for 3 h at 40-45 °C. The mixture was filtered,and the filtrate was diluted with water and extracted with diethyl ether. The extracts were dried over anhydrous Na2SO4 and the organic solvent was evaporated. The crude product was purified by flash column chromatography with EtOAc/hexane (1:2) as an eluent to obtain a white semi-solid 5a. Yield 49%; IR (cm-1): 2954,1720,1418,1369,1206; 1H NMR (400 MHz,CDCl3): δ 7.36- 7.25 (m,5H),5.63 (d,1H,J = 1.2 Hz),5.06 (d,1H,J = 15.2 Hz),4.83 (s,1H),4.21 (d,1H,J = 15.2 Hz),4.08 (d,1H,J = 1.3 Hz),1.57 (s,3H), 1.49 (s,3H); 13C NMR (100 MHz,CDCl3): δ 169.6,169.5,160.1, 135.3,128.8,129.2,127.9,71.3,67.0,60.6,58.3,52.9,52.0,46.3, 33.9,26.6; HRMS (FAB) calcd. for C19H22N3O5S2 [M+H]+: m/z 379.1327; found: 379.1334.
(3S,7R,7aR)-Dimethyl 6-(benzo[d]thiazol-2-ylmethyl)-2,2-dimethyl- 5-oxohexahydroimidazo[5, 1, b]thiazole-3,7-dicarboxylate (5b): Yield 40%; 1H NMR (400 MHz,CDCl3): δ 7.99 (d,1H, J = 8.2 Hz),7.89 (d,1H,J = 8.0 Hz),7.48 (t,1H,J = 8.0 Hz),7.43 (t, 1H,J = 8.0 Hz),5.71 (d,1H,J = 1.7 Hz),5.38 (d,1H,J = 16.6 Hz),4.83 (s,1H),4.77 (d,1H,J = 16.5 Hz),4.51 (d,1H,J = 1.7 Hz),3.81 (s,3H), 3.79 (s,3H),1.64 (s,3H),1.50 (s,3H); 13C NMR (100 MHz,CDCl3): d 169.3,166.7,159.7,153.0,135.4,126.3,125.4,123.2,121.9,71.2, 66.9,61.5,58.4,53.1,52.1,44.9,34.0,26.7; HRMS (FAB) calcd. for C18H22N3O5S2 [M+H]+: m/z 436.1001; found: 436.1014. 3. Results and discussion
3.1. Synthesis of thiazolidine amides through opening the β-lactam ring of penicillin V
Treatment of penicillin V with methyl iodide in DMF was carried out. Penicillin V methyl ester (1) as an intermediate was isolated and purified. With compound 1 available we developed an efficient procedure for generating a number of penicillin derivatives. Based on a parallel synthesis strategy,our method was to synthesize penicillin derivatives through opening the β-lactam ring of intermediate 1 with amines,which attacks the carbonyl group of β-lactam ring to form thiazolidine amides (Scheme 1). This synthetic approach was developed amenable to automation, enabling us to generate tens of compounds in a week using a Mettler Toledo MiniBlock Suite for library preparation and compound handling. In parallel,reaction of compound 1 with various aromatic,aliphatic and heterocyclic amines was carried out in six reactors,respectively. Six products (2a-2f) were produced simultaneously. They were purified and characterized by 1H NMR and 13C NMR spectra and HRMS.

|
Download:
|
| Scheme 1. Ring opening of β-lactams of penicillin V methyl ester. | |
{kind=link}
The rearrangement of the β-lactam ring started from 6-APA following our method (Scheme 2) modified from Johnson and Hardcastle [17]. Treatment of 6-APA with carbon dioxide was carried out in sodium bicarbonate aqueous solution. 6-APA was dissolved in water containing sodium bicarbonate. Carbon dioxide from a dry ice source was bubbled through the stirred mixture. The concentrated aqueous solution was then lyophilized to obtain the disodium salt of 8-hydroxypenillic acid in high yield. The 8- hydroxypenillic acid (isolated as the disodium salt by lyophilization) was characterized by 1H NMR and 13C NMR spectroscopy and HRMS. The NMR spectra of the disodium salt of 8-hydroxypenillic acid were well resolved and displayed no unassigned signals of significant intensity. The similar 1H NMR chemical shifts have been previously observed [18, 21]. Furthermore our 13C NMR chemical shifts are the same as the previous report [18, 21].

|
Download:
|
| Scheme 2. Synthesis of disodium salt of 8-hydroxypenillic acid from 6-APA. | |
{kind=link}
Using a parallel synthetic strategy,alkylation of 8-hydroxypenillic acid was initiated on MiniBlock by treatment of its disodium salt 3 with various aliphatic and aromatic halides as shown in Scheme 3. Disodium salt 3 was dissolved in DMF,and methyl iodide or benzyl bromide or propane iodide was added into the solution. The crude residues were purified to obtain pure products 4a-4c as white solids in moderate yields. Since N,N-diethylaminoethyl bromide hydrobromide was employed for the synthesis of 4d,excess sodium bicarbonate was added first into the solution to quench the hydrobromide acid. Compound 4d was isolated by extracting with diethyl ether from the water solution of the reaction mixture. Products (4a-4d) were confirmed by 1H and 13C NMR spectra and HRMS. In the 1H NMR spectra of compound 4b, two 2-proton multiplet peaks of methylene groups at δ 4.17 were indicative of the formation of propyl esters at C3 and C6 positions. The complex patterns at δ 7.38 and 5.16 affirmed the presence of benzyl methyl esters in the compound 4c. For product 4b,the patterns at δ 4.25,4.17,2.74,2.60,0.97 confirmed the formation of diethylaminoethyl esters.

|
Download:
|
| Scheme 3. Synthesis of 8-hydroxypenillic acid esters. | |
{kind=link}
Furthermore,alkylation of 8-hydroxypenillic acid methyl ester (4a) was carried out at its N-7 position with benzyl bromide or 2- (bromomethyl)benzo[d]thiazole using potassium carbonate as base in DMF (Scheme 4). Benzyl bromide and anhydrous potassium carbonate were added to a solution of 4a in DMF and the mixture was stirred. The crude product was purified to obtain a pure product 5a as a white semi-solid in modest yield. Similar procedure was proceeded for the synthesis of 5b by using 2- (bromomethyl)benzo[d]thiazole as an alkylation reagent. Compounds 5a and 5b were identified by 1H NMR and 13C NMR and HRMS. In the 1HNMRspectra of compound 5a,the chemical shift of the methylene in the benzyl group supported the benzyl alkylation at N-7 position,and the signals from its β-lactam protons were identical in position and pattern with those in the spectrum of the starting penillic acid methyl ester 4a. For product 5b,the complex patterns of benzo[d]thiazol-2-ylmethyl group confirmed the achievement of alkylation at N-7 position by 2-(bromomethyl)- benzo[d]thiazole.

|
Download:
|
| Scheme 4. Alkylation of 8-hydroxypenillic acid methyl ester at N-7 position. | |
{kind=link}
Chemical arrangements of penicillin β-lactam ring were undertaken in this work. Six thiazolidine amides were synthesized through N4-C7 β-lactam ring opening of penicillin V methyl ester with various aliphatic,aromatic,and heterocyclic primary amines. Five 8-hydroxypenillic acid derivatives with side chains of methyl, propyl,benzyl,diethylaminoethyl,and 2-(bromomethyl)benzo[ d]thiazole groups were yielded via β-lactam ring rearrangement from 6-aminopenicillanic acid (6-APA). Parallel synthetic methods were developed for esterification of 8-hydroxypenillic acid and β-lactam ring opening of penicillin V methyl ester. The compounds display biological activities.
| [1] | K.B. Hoten, E.M. Onusko, Appropriate prescribing of oral β-lactam antibiotics, Am. Fam. Physician 62 (2000) 611-620. |
| [2] | G. Lukacs, Recent Progress in the Chemical Synthesis of Antibiotics and Related Microbial Products, vol. 2, Springer, Berlin, 1993p. 621. |
| [3] | G. Veinberg, M. Vorona, M. Shestakova, I. Kanepe, E. Lukevics, Design of β-lactams with mechanism based nonantibacterial activities, Curr. Med. Chem. 10 (2003) 1741-1757. |
| [4] | R.R. Gupta, Topics in Heterocyclic Chemistry, Springer, Berlin/Heidelberg, 2010p. 394. |
| [5] | J.D. Rothstein, S. Patel, M.R. Regan, et al., b-Lactam antibiotics offer neuroprotection by increasing glutamate transporter expression, Nature 433 (2005) 73-77. |
| [6] | P.G. Sammes, Recent chemistry of the β-lactam antibiotics, Chem. Rev. 76 (1976) 113-155. |
| [7] | A.K. Mukerjee, A.K. Singh, b-Lactams: retrospect and prospect, Tetrahedron 34 (1978) 1731-1767. |
| [8] | G.H. Hakimelahi, K.S. Shia, C. Xue, et al., Design, synthesis, and biological evaluation of a series of β-lactam-based prodrugs, Bioorg, Med. Chem. 10 (2002) 3489- 3498. |
| [9] | T.L. Gilchrist, Heterocyclic Chemistry, 2nd ed., Longman Press, Harlow, 1997. |
| [10] | S. Wolfe, S. Ro, C.K. Kim, Z. Shi, Synthesis and decarboxylation of Δ2-cephem-4 4- dicarboxylic acids, Can. J. Chem. 79 (2001) 1238-1258. |
| [11] | B. Alcaide, P. Almendros, C. Aragoncillo, b-Lactams: versatile building blocks for the stereoselective synthesis of non β-lactam products, Chem. Rev. 107 (2007) 4437-4492. |
| [12] | B. Alcaide, P. Almendros, G. Cabrero, M.P. Ruiz, Stereoselective cyanation of 4- formyl and 4-imino-b-lactams: application to the synthesis of polyfunctionalized γ-lactams, Tetrahedron 68 (2012) 10761-10768. |
| [13] | B. Alcaide, P. Almendros, A. Luna, et al., Controlled rearrangement of lactamtethered allenols with brominating reagents: a combined experimental and theoretical study on alpha-versus β-keto lactam formation, Chem. Eur. J. 17 (2011) 11559-11566. |
| [14] | S. Dekeukeleire, M. D'hooghe, N. De Kimpe, Diastereoselective synthesis of bicyclic gamma-lactams via ring expansion of monocydic β-lactams, J. Org. Chem. 74 (2009) 1644-1649. |
| [15] | W. Van Brabandt, N. De Kimpe, Electrophile-induced ring expansions of β-lactams toward γ-lactams, J. Org. Chem. 70 (2005) 8717-8722. |
| [16] | A.K. Mukerjee, A.K. Singh, Reactions of natural and synthetic β-lactams, Synthesis- Stuttgart 9 (1975) 547-589. |
| [17] | D.A. Johnson, G.A. Hardcastle Jr., Reaction of 6-aminopenicillanic acid with carbon dioxide, J. Am. Chem. Soc. 83 (1961) 3534-3535. |
| [18] | R.F. Pratt,M. Dryjanski, E.S. Wun, V.M. Marathias, 8-Hydroxypenillic acid from 6- aminopenicillanic acid: a new reaction catalyzed by a class C β-lactamase, J. Am. Chem. Soc. 118 (1996) 8207-8212. |
| [19] | C.C. Ruddle, T.P. Smyth, Exploring the chemistry of penicillin as a β-lactamasedependent prodrug, Org. Biomol. Chem. 5 (2007) 160-168. |
| [20] | L. Heinisch, S. Wittmann, T. Stoiber, et al., Highly antibacterial active aminoacyl penicillin conjugates with acylated bis-catecholate siderophores based on secondary diamino acids and related compounds, J. Med. Chem. 45 (2002) 3032- 3040. |
| [21] | A.F. Casy, A. Lipczynski, 8-Hydroxypenillic acid: NMR characteristics and facile formation from 6-aminopenicillanic acid, J. Pharm. Pharmacol. 46 (1994) 533-534. |
| [22] | PubChem: https://pubchem.ncbi.nlm.nih.gov. The compound identification (CID) numbers are 24747544 (2a), 24747436 (2b), 24747499 (2c), 24747495 (2d), 24747364 (2e), 24747549 (2f), 25011528 (3), 25011526 (4a), 24747497 (4b), 24747437 (4c), 25011527 (5a), 24789294 (5b). |