




b Key Laboratory for Green Pharmaceutical Technologies and Related Equipment of Ministry of Education, College of Pharmaceutical Sciences, Zhejiang University of Technology, Hangzhou 310014, China
Imidodicarbonic diamides (Fig. 1) exhibit a wide range of biological activities,such as anti-tumor and antioxidation [1, 2, 3, 4]. Furthermore, they play akeyrole inthe synthesisofmacrocyclicnitrogen-containing compounds such as mononitrobiuret (MNB) and 1,5-dinitrobiuret (DNB) [5, 6, 7, 8]. In general,they can be obtained froma certain purines by oxidation or decomposition [9, 10]. However,the synthesis of these compounds has received very little attention.

|
Download:
|
| Fig. 1. Structures of imidodicarbonic diamide,MNB and DNB. | |
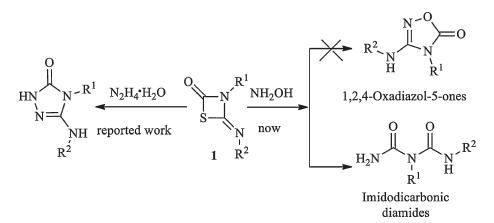
It is well known that the four-membered heterocycle 4-imino- 1,3-thiazetidin-2-ones 1 are useful organic intermediates in organic synthesis. Recently,our group [11] reported the ringopening reaction of 1 with N2H4·H2O (Scheme 1). As an extension, herein,we wish to study the ring-opening reaction in the presence of hydroxylamine. The experiment results showed the title products were imidodicarbonic diamides 2 rather than 1,2,4- oxadiazol-5-ones.

|
Download:
|
| Scheme 1. Ring-opening reaction of 1 with N2H4·H2O or NH2OH·HCl. | |
Analytical grade solvents and commercially available reagents were used without further purification. The flash column chromatography was carried out over silica gel (200-400 mesh), purchased from Qingdao Haiyang Chemical Co.,Ltd. Melting points were determined on a Buchi B-540 capillary melting point apparatus and uncorrected. IR spectra was recorded on an AVATAR-370,samples were prepared as KBr plates. 1H NMR and 13C NMR spectrawere recorded at VARAIN-400 using CDCl3 as the solvent with tetramethylsilane (TMS) as an internal standard. Chemical shifts are given in δ relative to TMS,the coupling constants J are given in Hz. Mass spectra were measured with Thermo Finnigan LCQ-Advantage. High resolution mass spectral (HRMS) analyze were measured on an Agilent 6210 TOF LC/MS using ESI or EI (electrospray ionization) techniques. The X-ray data was recorded on Gemini Ultra EXXH-236/09.
Syntheses of 4-(arylimino)-1,3-thiazetidin-2-ones 1a-1j: To a mixture of ethyl acetate (25 mL),sodium bicarbonate (11.5 mmol, 0.97 g) and thioureas (5 mmol),BTC [bis(trichloromethyl)carbonate] (1.7 mmol,0.5 g) was added carefully in portions. The reaction mixture was stirred at room temperature for 15 min. After completion (by TLC),the mixture was filtered,the organic solvent was condensed and the compounds 1 were obtained as crystal almost quantitative.
Syntheses of imidodicarbonic diamides 2a-2h and 3f-3h: 3- Aryl-4-(arylimino)-1,3-thiazetidin-2-ones 1 (1 mmol) were added into a mixture of hydroxylamine hydrochloride (1.2 mmol) and imidazole (1.2 mmol) in ethanol (15 mL). The reaction would last about 25 min at room temperature and the crude product was purified by flash chromatography (ethyl acetate/petroleum ether, 1/6) to provide the pure product.
Syntheses of 1-alkyl-1-arylureas 4i and 4j: 1 (1.0 mmol) were added into a mixture of hydroxylamine hydrochloride (1.2 mmol) and imidazole (1.2 mmol) in ethanol (15 mL). The reaction would last about 3 h at room temperature and the crude product was purified by flash chromatography (ethyl acetate/petroleum ether, 1/8) to provide the pure product.
Syntheses of 1-carbamothioyl-1-phenylureas 5f and 5j: 1 (1.0 mmol) were added into a mixture of hydroxylamine hydrochloride (1.2 mmol) and pyridine (1.2 mmol) in methanol (15 mL). The reaction would last about 2 h at room temperature and the crude product was purified by flash chromatography (ethyl acetate/petroleum ether,1/6) to provide the pure product.
Physical and chemical data of the chosen product is demonstrated below.
1-Carbamoyl-1,3-bis(4-chlorophenyl)urea (2a): White solid; mp: 177.6-179.8 °C. IR (KBr,cm-1): vmax 3489,3172,1681,1596, 1404. 1H NMR (400 MHz,CDCl3): δ 10.73 (s,1H),7.49 (d,2H, J = 8.8 Hz),7.40 (d,2H,J = 8.8 Hz),7.28 (d,2H,J = 8.8 Hz),7.23 (d, 2H,J = 8.8 Hz),5.18 (s,2H). 13C NMR (100 MHz,CDCl3): δ 156.7, 151.8,136.0,135.6,135.2,130.6 (CH × 2),130.4 (CH × 2),128.9, 128.8 (CH × 2),121.2 (CH × 2). MS (ESI): m/z 346 [M+Na]+. HRMSESI: calcd. for C14H11Cl2N3NaO2: 346.0126; found: 346.0130.
1-Carbamoyl-1-(4-chlorophenyl)-3-phenylurea (3f): White solid; mp: 144.7-147.5 °C. IR (KBr,cm-1): vmax 3482,3221, 1677,1602,1446. 1H NMR (400 MHz,CDCl3): δ 10.36 (s,1H),7.48 (d,2H,J = 8.4 Hz),7.43 (d,2H,J = 8.4 Hz),7.31-7.26 (m,4H),7.07 (t, 1H,J = 7.6 Hz),5.31 (s,2H). 13C NMR (100 MHz,CDCl3): δ 157.3, 152.3,137.1,136.5,130.4 (CH × 2),129.7,129.4 (CH × 2),129.0 (CH × 2),128.9,121.4 (CH × 2). MS (ESI): m/z 312 [M+Na]+. HRMSESI: calcd. for C14H12ClN3NaO2: 312.0516; found: 312.0527.
1-Isopropyl-1-phenylurea (4i): White solid; mp: 154.9- 159.5 °C. IR (KBr,cm-1): ymax 3347,3187,1693,1555,1442. 1H NMR (400 MHz,CDCl3): δ 7.26 (d,4H,J = 7.2 Hz),7.02-7.05 (m, 1H),6.70 (s,1H),3.97 (m,1H),1.15 (dd,6H,J1 = 2.0 Hz,J2 = 6.4 Hz). 13C NMR (100 MHz,CDCl3): δ 138.8,129.3 (CH × 2),123.6,120.9 (CH × 2),42.4,23.6 (CH × 2). MS (ESI): m/z 179 [M+1]+. HRMS-ESI: calcd. for C10H15N2O: 179.1184; found: 179.1186.
1-(4-Chlorophenyl)-1-(phenylcarbamothioyl)urea (5f): White solid; mp: 117.6-120.1 °C. IR (KBr,cm-1): ymax 3435,3265,1702, 1534,1033. 1H NMR (400 MHz,CDCl3): δ 12.46 (s,1H),7.56 (d,2H, J = 7.6 Hz),7.41-7.36 (m,4H),7.26 (d,1H,J = 7.2 Hz),7.14 (dd,2H, J1 = 2.0 Hz,J2 = 7.6 Hz),3.75 (s,3H,CH3). 13C NMR (100 MHz, CDCl3): δ 182.0,156.2,140.0,136.9,131.8,130.5,129.1 (CH × 2), 128.8,128.7,128.2,126.2 (CH × 2),125.0,54.6. MS (ESI): m/z 321 [M+1]+. HRMS-ESI: calcd. for C15H14ClN2O2S: 321.0465; found: 321.0465. 3. Results and discussion
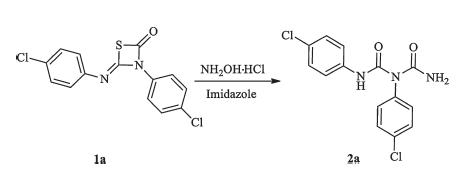
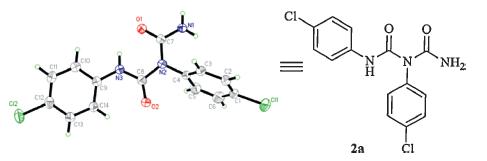
3-(4-Chlorophenyl)-4-((4-chlorophenyl)imino)-1,3-thiazetidin- 2-one 1a was selected as the starting material for the ringopening reaction in our initial studies,which was synthesized from thiourea with triphosgene in excellent yield (up to 95%) [12]. Then 1a was treated with hydroxylamine hydrochloride in the presence of imidazole under reflux conditions for 35 min. It was found that 1a reacted with hydroxylamine quickly with the release of H2S (Scheme 2). A novel compound 2a was gotten in 88% yield. The structure 2a was confirmed via single crystal X-ray diffraction analysis (Fig. 2,details see Supporting information).

|
Download:
|
| Scheme 2. Unexpected 2a was synthesized by 1a. | |

|
Download:
|
| Fig. 2. The X-ray structure of 2a. | |
Then,we focused on screening for the reaction conditions promoted by these results in mild conditions. In order to minimize the generation of side products that could arise from the reaction between substrate 1a and organic base imidazole,the procedure was firstly proceeded in the presence of equal equivalent of imidazole and NH2OH·HCl before the addition of 1a. Experiment results showed that the solvents had great influence on this reaction (Table 1). Solvents such as ethanol and CH2Cl2,had little effect on the yields of 2a (Table 1,entries 2-4). None of 2a (Table 1, entry 1) was observed by using water as solvent. Hence,ethanol was selected as solvent and imidazole (pKa = 14.5) was chosen as the base to free NH2OH·HCl (pH 6.5) at room temperature [13, 14, 15, 16].
With the determined structure of the product and the optimized reaction conditions in hand,we then turned to explore the scope of this reaction. It showed that 1 reacted with hydroxylamine smoothly,and two products 2 (minor) and 3 (major) were obtained after 25 min (Scheme 3).
| Table 1 Preparation of 2a from 1a in different solventsa |
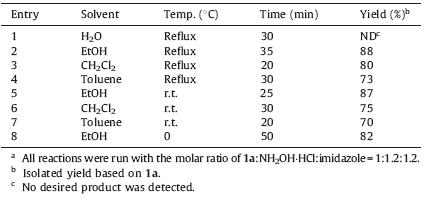

|
Download:
|
| Scheme 3. Regioselective synthesis of 3 from 1. | |
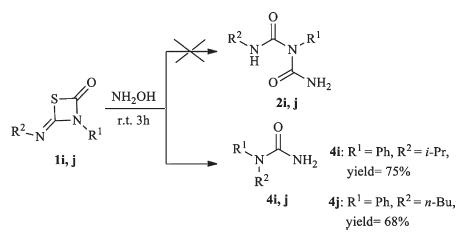
In order to investigated further similar reactions,various 1,3- thiazetidin-2-ones were subsequently used as the substrates to study the scope and general applicability of the protocol,as shown in Table 2. In most cases,the desired products were gotten in good yields (Table 2,76%-89%). When symmetrical thiourea (R1 = R2) was used in this reaction,the only product (2 = 3) was gotten in the presence of NH2OH. Then,we turned to study the effects of substrates by using unsymmetrical thiourea (R1 ≠ R2). It was found that compounds 1 with electron-donating groups showed higher selectivity than these with electron-withdrawing group on benzene ring (Compounds 1f,1g and 1h). The regioselectivity of this ring-opening reaction may be correlated with the electron density of the substituents on the aromatic rings. Moreover, substituents with bulky groups on the aromatic rings showed a dramatic effect on the formation of 2 (compounds 1g). The higher steric hindrance they had,the lower yields of 2 were obtained. When alkyl groups were investigated in this reaction,none of the expected compounds 2 and 3 were obtained (Compounds 1i and 1j). The 1,3-δ-shift reaction occurred,and products of 1-isopropyl- 1-phenylurea 4i and 1-butyl-1-phenylurea 4j were in 75% and 68% yield,respectively (Scheme 4).
| Table 2 Synthesis of imidodicarbonic diamides 2 and 3 from 4-imino-1,3-thiazetidin-2- ones 1a |
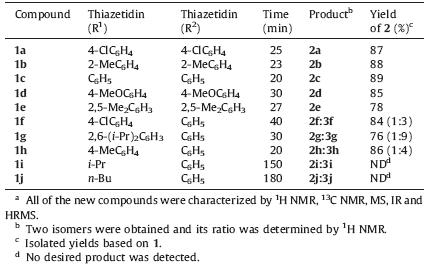
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}

|
Download:
|
| Scheme 4. Synthesis of 4i and 4j. | |
{kind=link}
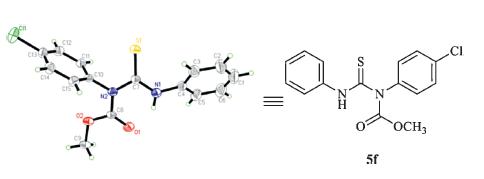
During the synthesis of imidodicarbonic diamide,we found another experiment phenomenon. When methanol and pyridine were used,the reaction could also proceed in 2 h.However,wecould not obtain the product 4f,while another compound thiourea 5f was gotten in 47% yield (Scheme 5). The crystal structure of compound 5f was shown in Fig. 3 (details see Supporting information).
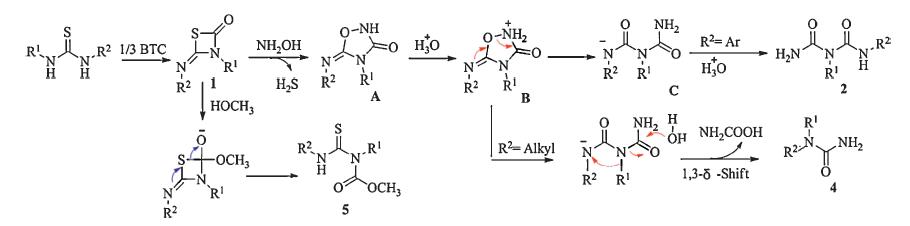
On the basis of the above experimental results,a possible mechanism is shown in Scheme 6 [17, 18]. Intermediates 1 firstly reacted with NH2OH to form A and H2S. Then,an intramolecular ring-opening reaction took place to afford transition state B in the presence of H+,followed by addition of proton to afford the imidodicarbonic diamides 2 (R2 = Ar). When R2 is alkyl group,B further underwent in situ 1,3-d-shift reaction to yield 4. Besides, compound 5 can be obtained by nucleophilic addition of CH3OH to compound 1.

|
Download:
|
| Scheme 5. The preparation of 5f. | |
{kind=link}

|
Download:
|
| Scheme 6. Proposed reaction mechanism. | |
{kind=link}

|
Download:
|
| Fig. 3. X-ray structure of 5f. | |
{kind=link}
In summary,a novel and efficient route for the construction of imidodicarbonic diamides was described,with the advantages of straightforward procedure,mild conditions,good yields,simple operation,and easily available materials. Furthermore,in similar conditions,two different scaffolds were obtained selectively in good yields. In addition,the unexpected compounds were confirmed by X-ray analysis.
AcknowledgmentsWe are grateful to the Natural Science Foundation of Zhejiang Province (No. LY13B020015) and the Science and Technology Plan Project of Taizhou (No. 131KY03) for financial support.
Appendix A. Supplementary dataSupplementary data associated with this article can be found, in the online version, at http://dx.doi.org/10.1016/j.cclet.2014.10.015.
| [1] | G.N. Rozentsveig, A.V. Popov, I.B. Rozentsveig, G.G. Levkovskaya, Reactions of N- (2,2,2-trichloroethylidene)- and N-(2,2-dichloro-2-phenylethylidene)arenesulfonamides with biuret, Russ. J. Org. Chem. 44 (2008) 1486-1489. |
| [2] | C. Boss, M.H. Bolli, T. Weller, W. Fischli, M. Clozel, Bis-sulfonamides as endothelin receptor antagonists, Bioorg. Med. Chem. Lett. 13 (2003) 951-954. |
| [3] | A. Sakakura, K. Suzuki, H. Katsuzaki, et al., Hanasanagin: a new antioxidative pseudo-di-peptide, 3,4-diguanidinobutanoyl-DOPA, from the mushroom, Isaria japonica, Tetrahedron Lett. 46 (2005) 9057-9059. |
| [4] | Y.B. Chen, J.L. Li, X.S. Shao, X.Y. Xu, Z. Li, Design, synthesis and insecticidal activity of novel anthranilic diamides with benzyl sulfide scaffold, Chin. Chem. Lett. 24 (2013) 673-676. |
| [5] | L. Kou, J. Liang, X.H. Ren, et al., Novel N-halamine silanes, Colloids Surf. A: Physicochem. Eng. Asp. 345 (2009) 88-94. |
| [6] | H. Ito, K. Yumura, K. Saigo, Synthesis, characterization, and binding property of isoelectronic analogues of nucleobases, B(6)-substituted 5-aza-6-borauracils and -thymines, Org. Lett. 12 (2010) 3386-3389. |
| [7] | M. Roman, B. Andrioletti, M. Lemaire, et al., Investigations providing a plausible mechanism in the hexamethyldisilazane-catalyzed trimerization of alkyl isocyanates, Tetrahedron 67 (2011) 1506-1510. |
| [8] | Y. Sanemitsu, M. Shiroshita, K. Maeda, S. Inoue, A novel class of fungicides: 2H- 1,2,4,6-thiatriazine-3,5(4H,6H)-dione-1-oxides, Agric. Biol. Chem. 51 (1987) 3173-3175. |
| [9] | R.R. Oltjen, E.E. Williams Jr., L.L. Slyter, G.V. Richardson, Urea versus biuret in a roughage diet for steers, J. Anim. Sci. 29 (1969) 816-822. |
| [10] | F.J. Moore, E.S. Gatewood, The action of hydrogen peroxide upon certain phenylsubstituted uric acids, J. Am. Chem. Soc. 45 (1923) 135-145. |
| [11] | C. Jin, C.W. Liu, W.K. Su, Novel synthesis of 2,4-dihydro-5-amino[1,2,4]triazol-3- ones from 1,3-disubstituted thioureas, Synlett (2009) 607-610. |
| [12] | I.J. Enyedy, J. Wang, W.A. Zaman, K.M. Johnson, S. Wang, Discovery of substituted 3,4-diphenyl-thiazoles as a novel class of monoamine transporter inhibitors through 3-D pharmacophore search using a new pharmacophore model derived from mazindol, Bioorg. Med. Chem. Lett. 12 (2002) 1775-1778. |
| [13] | K. Okuda, Y.X. Zhang, H. Ohtomo, T. Hirota, K. Sasaki, Polycyclic N-heterocyclic compounds. Part 62: reaction of N-(quinazolin-4-yl)amidine derivatives with hydroxylamine hydrochloride and anti-platelet aggregation activity of the products, Chem. Pharm. Bull. 58 (2010) 369-374. |
| [14] | C.L. Carroll, A.R. Chamberlin, Synthesis of the dysiherbaine tetrahydrophran core utilizing improved tethered aminohydroxylation conditions, Tetrahedron Lett. 52 (2011) 3995-3997. |
| [15] | V.Y. Sosnovskikh, V.S. Moshkin, R.A. Irgashev, Reactions of 3-(polyfluoroacyl)- chromones with hydroxylamine. The first synthesis of 3-cyano-2-(polyfluoroalkyl) chromones, Tetrahedron Lett. 47 (2006) 8543-8546. |
| [16] | S. Man, M. Buchlovič, M. Potáček, Cyclization of β-allenyloximes as a novel method for nitrone preparation, Tetrahedron Lett. 47 (2006) 6961-6963. |
| [17] | C. Pérez-Balado, B. Iglesias, L. Muñoz, Different reactivity of hydroxylamine with carbamoyl azides and carbamoyl cyanides: synthesis of hydroxyureas and carbamoyl amidoximes, J. Org. Chem. 75 (2010) 8039-8047. |
| [18] | N. Poje, M. Poje, An unusual oxidative ring transformation of purine to imidazo[ 1,5-c]imidazole, Org. Lett. 5 (2003) 4265-4268. |