



 , Rui Wang
, Rui Wang
Peroxisome proliferator activated receptor γ (PPARγ )isa subtype of peroxisome proliferator activated receptors (PPARs) family,and plays an important role in improving insulin sensitivity and regulating the storage and catabolism of dietary fat [1,2,3]. As a member of the nuclear receptor gene family,PPARγ is expressed in many tissues,especially in skin,brain,and adipose tissue,and regulates the gene expression in response to the ligand binding [2,4]. Recent studies have also demonstrated PPARγ is an efficient target to develop agonists for therapies of metabolic diseases,such as the type II diabetes,atherosclerosis and inflammatory skin disease [5,6,7,8,9,10,11,12,13]. Furthermore,PPARγ agonists also have exerted neuroprotective effect against oxidative-stress induced apoptosis, which can be used in chronic neurodegenerative disorders including Alzheimer’s and Parkinson’s disease [14].
Thiazodidiendiones (TZDs) were the first class of high-affinity synthetic PPARγ agonists,some of which have been used as antidiabetic drugs in recent clinical studies [15,16,17,18,19,20]. Among them, Pioglitazone,Rosiglitazone and Troglitazone are the classic TZDs drugs for antidiabetic treatments,but Troglitazone was withdrawn from the market owing to the severe side effect of liver damage [21,22]. Recently,the anti-inflammatory and neuroprotective effects of PPARγ agonists have also attracted wide attention in the various types of neurodegenerative disorders. There is considerable evidence that PPARγ agonists regulate the microglial mediated inflammatory responses and inhibit the expression of proinflammatory gene. PPARγ agonists inhibit the expression of inflammatory cytokines,MMPs,COX-2,TNF-a,iNOS by attenuating nuclear factor kB (NF-kB) activation [23,24,25]. Glass and colleagues reported that PPARγ represses NF-kB target genes dependent inflammatory responses [26]. Feinstein’ study demonstrated that Pioglitazone suppressed iNOS and COX-2 expression in the AD brains [27]. Moreover,Rosiglitazone significantly improved the cognitive deficiency in AD patients and animal models [28]. Unfortunately,Rosiglitazone has been removed from the market as well,which can cause heart attack and is risky for patients to take, especially the ones with a history of heart diseases [29,30]. Recently,Pioglitazone turns to be the most common and popular antidiabetic agent in the market,while it is still suggested to be associated with the possible risk to induce bladder tumors [31,32,33]. Therefore,there is a growing need for developing more potent agents acting on PPARγ to ensure their clinical applicability and decrease unwanted side effects.
Moreover,abundant investigations on TZDs [34] indicated us to keep the TZD and phenyl ring,which are critical parts to interact with the receptor in the PPARγ ligand binding domain (LBD). Importantly,the structure of the PPARγ LBD bond to the TZD Rosiglitazone together has been solved,which revealed that the Rosiglitazone only occupies 40% of the PPARγ ligand binding site in a U-shaped binding conformation,while the rest space of pocket of PPARγ receptor remains unoccupied [5].
With these in mind,we designed and synthesized a series of 2-thioxothiazoline-4-one analogs and evaluated them on PPARγ binding activities. Each of these derivatives has a 2-thioxothuazoline-4-one analog as the head group,the phenyl ring analogs of terminal,and an unsaturated linker combining the above two analogs which makes the molecules stiffer and more resistant to twisting and thus could lead to different conformations occupying other space of the LBD. Structure-activity relationship (SAR) was studied to find out superior substituent alternatives to this series. Then molecular fitting of these compounds onto the approved drug Rosiglitazone in the PPARγ ligand binding domain was taken to elucidate the SAR and explore potential receptor-ligand interactions. 2. Experimental 2.1. General methods
All reagents and solvent were commercial quality without further purification.
1H NMR and 13C NMR were measured on a Bruker AV-400 spectrometer using TMS as internal standard. The chemical shifts were reported as ppm (in DMSO-d6). Melting points were measured with an X6 apparatus,which were uncorrected. High-resolution EI mass spectra were reported with a HPLC-Q-Tof MS (Micro) spectrometer. All the compounds were visualized by UV illumination (254 nm). Analytical thin-layer chromatography was performed with glass-baked silica gel plates. HPLC was performed using a Zopbax RX-C18 column (packing: 5mm, 4.6 mm × 150 mm) with a mobile phase consisting of H2O/CH3CN at a flow rate of 1.0 mL/min.
General procedure and characterization data for the preparation of compounds (9,30-33,35-40,42,44) in Table 1 and other compounds (a6-a14,a16,a18,a19,a21,a23-a26,a28,a29,a31, a32) in Table S1 are detailed in the Supporting information.
|
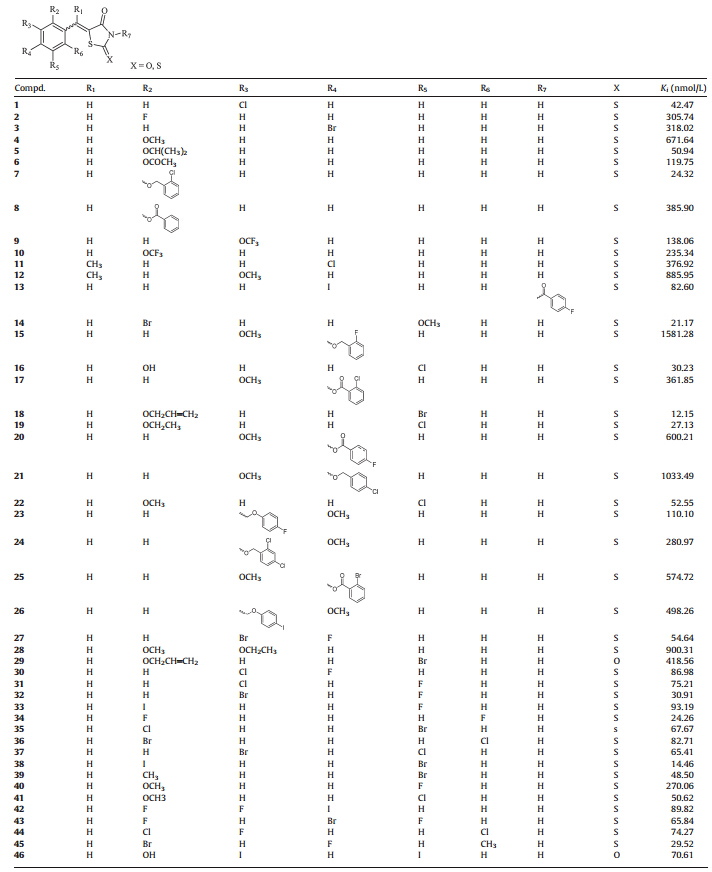
Table 1 Compounds 1-46 with PPARγ binding activities. |
The compounds targeted to PPARγ were screened using PPARγ amma Competitive binding assay kit (Invitrogen) [35]. Briefly,compounds (diluted from 10-2 mol/L dimethylsulfoxide stocks to 10-4 -10-11 mol/L),fluormone Pan-PPAR Green (the acceptor,20 nmol/L),PPARγ -LBD/Tb-anti-GST Ab (the donor, 20 nmol/L) in complete TR-FRET PPAR Assay buffer (TR-FRET PPAR Assay buffer containing 5 mmol/L DTT) were added to each well of a black flat-bottom 384-well polystyrene microplates (Corning),incubated for 6 h at room temperature. TR-FRET signals were measured using Enspire Multimode Reader (excitation: 340 nm,acceptor-emission: 520 nm,donor-emission: 495 nm). The data were calculated as the ratio of the emission intensity of acceptor and donor fluorophores. The inhibitor constant (Ki) for a competitor can be calculated by applying the Cheng-Prusoff equation [36] (Cheng & Prusoff,1973):Ki =IC50/(1 + ([tracer]/KD)), Tracer = 5 nmol/L,KD= 2.8±0.8 nmol/L. 2.3. Computational methods
To save computational time and resources,we performed molecular superposition simulations firstly for the 91 compounds using a rapid 3D molecular similarity calculating software,SHAFTS [37]. All compounds were stored in a 2D SDF file and an original conformation would be generated for each compound,then a conformation generation program based on a multi-objective evolution algorithm,Cyndi,was used to generate the conformation ensembles (100 ones as default) for each compound [38]. The template structure was the crystal conformation of an approved PPARγ agonist Rosiglitazone bound to the ligand binding domain (LBD) of PPARγ with the PDB code of 2PRG [39]. Then SHAFTS would do the molecular fittings considering both the shape factor and the chemotype factor to build more valid models. Moreover, we utilized MacroModel in Maestro (Schro¨dinger,Version 7.5) on further conformation optimizations with default settings for each compound in the LBD of PPARγ (2PRG) to refine potential sterical clash to residues of the protein by molecular fittings aforementioned. 3. Results and discussion 3.1. Synthetic strategy
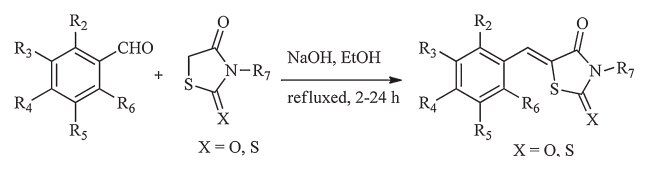
The compounds (a1-a34) in Table S1 can be found in the Supporting information. The synthetic route used to obtain the compounds (9,30-33,35-40,42,44) and numbers of compounds (a6-a14,a16,a18,a19,a21,a23-a26,a28,a29,a31,a32) are displayed in Scheme 1,which was achieved by previously reported methods [40]. Other compounds listed in Table 1,Table S1 and Table 2 were purchased from a commercial reagent company. Generally speaking,2-thioxo-4-thiazolidinones and various commercially available benzaldehydes were added together to ethanol in the presence of sodium hydroxide,the mixture was heated to reflux for 2-24 h. After a simple extraction,the resulting residue was purified by recrystallization from ethanol to get the final product.

| Download: |
| Scheme 1.Synthesis of 2-thioxo-4-thiazolidinone derivatives. | |
|
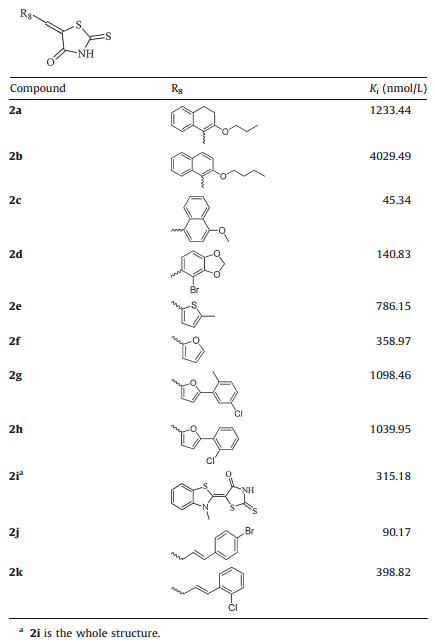
Table 2 Compounds 2a-2 kwith PPARγ binding activities. |
{kind=link}
As illustrated in Fig. 1,we retained the basic TZD scaffold with certain simple modifications and considered more substitutions on the linker and the terminal phenyl ring. Firstly,the thiocarbonyl group was chosen to replace the carbonyl group in the 2-position of TZD and an unsaturation was introduced to the linker together to generate a novel TZD scaffold,then certain common substituent groups on the scaffold were considered for investigations at the same time. Secondly,the substitutions were introduced along with the linker,and the length of the linker was also considered. Then various substituted groups were introduced onto the phenyl ring to attain a balance of the PPARγ binding activity and the structural diversity.

| Download: |
| Fig. 1. The structural modification progress of 2-thioxo-4-thiazolidinone derivatives. | |
{kind=link}
Both the synthesized compounds and the commercial purchased ones were evaluated by the binding activities of PPARγ ,and the structure activity relationship (SAR) was discussed with the binding constant (Ki) values. As a whole,28 (1,5,7,13,14,16,18, 19,22,27,30-39,41-46,2c,2j) compounds exhibited moderate binding activities withKi values less than 100 nmol/L,and 9 compounds (7,14,16,18,19,32,34,38,45) showed excellent binding activities withKi values less than 31 nmol/L,especially the most potent compound18with the Ki value of 12.15 nmol/L. 3.2.1. Modification of the TZD scaffold
As discussed above,modifications were first focused on the scaffold. Replacing the carbonyl in the 2-position of TZD ring of compound 29 to a thiocarbonyl generated 18,which could reach a 34-fold improvement in the binding activity. Then introducing hydrophobic group to replace the critical hydrogen bond of the N-position of TZD ring,the acylation of the N atom of compound 13 still retain the binding activity (Ki = 82.60 nmol/L). These results suggested that the thiocarbonyl at the 2-position of TZD can significantly improve the binding activity. Thus the 2-thioxo-4-thiazolidinone is the optimum head group,whilst substitutions to the N atom of the TZD can be tolerated. 3.2.2. Modification of the linker
Extending the unsaturated linker between the 2-thioxo-4-thiazolidinone and phenyl ring from one carbon unit (3) to three (2j,in Table 2) resulted in a 3-fold improvement in the PPARγ binding activity with theKi value of 90.17 nmol/L,while the compound 2k with an extended linker did not improve the activity. Besides,we also noticed that introducing the methyl group (11 and 12) to the unsaturated linker did not improve the activity. These results demonstrated that substituents on the phenyl ring play a more important role in keeping binding activities,while the linker between the TZD and phenyl ring could be extended to more than one carbon unit. Meanwhile the substitutions within the unsaturation linker also can be tolerated. 3.2.3. Modification of the phenyl ring
Thus with the 2-thioxo-4-thiazolidinone scaffold and the unsaturation linker unchanged,we moved on to introduce common substituent groups to the phenyl ring. Consequently comparing 4,5,6,8 with 7 in Table 1,which were all monosubstituted at the 2-position of phenyl ring,it was observed that replacing the methoxy group (4) with the isopropoxy group (5) resulted in a 13-fold improvement in the PPARγ binding activity. The 4-cloride benzyloxy derivative (7) evenshowedasuperior binding activity with the Ki value of 24.32 nmol/L,while the acylation derivatives 6 and 8 led to a slight improvement in the binding activity to the compound 4. But,introduction of an ethoxy group to the 3-position of the phenyl ring in 4 furnished compound 28 could attenuate the activity. Moreover,introducing a Cl group to the 5 position of the phenyl ring in 4 furnished compound 22 could increase the binding activity by 12-fold with Ki value of 52.55 nmol/L,while the replacement of the Cl atom of 22 with a F atom (40) resulted in a 5-fold loss in binding activity (Ki= 270.06 nmol/L). With these in mind,we demonstrated that a lot of substituent groups could be tolerated at the 2-position of the phenyl ring,especially mono-substituted larger hydrophobic groups apparently benefited from binding activities,while halogen atoms such as Cl and Br were preferred at the 5-position as further modifications.
For the 3-position substituent of the phenyl ring,we found that any further modifications at other positions of the phenyl ring,with the Cl atom substituents unchanged (1),could not improve the activity. Additionally,comparing compounds 31 with 32,it was demonstrated that a larger halogen atom at the 3-position of the phenyl ring was more appropriate for structural optimization. For the 4-position substituent of the phenyl ring, compound 3 with the monobromo at the 4-position of the phenyl ring had moderate binding activity (Ki=318.02nmol/L). Furthermore,multi-substitutions were introduced to the phenyl ring of compound 3 for derivatization. We found that introducing F atoms to both 2 and 5 position of phenyl ring of compound 3 resulted in a roughly 5-fold improvement in binding activity (compound 43,Ki= 65.84 nmol/L),while other multi-substituted derivatives did not show the obvious improvement of activities. Besides,substituted phenyl rings were introduced to the 4-position of terminal phenyl ring to investigate the space tolerance. When compared to compounds 15,17,20,21,25,the esterification of the hydroxy group at the 4-position of the phenyl ring with benzoate derivatives were superior to etherification with benzyl derivatives in the improvement of binding activity. These observations indicated that various hydrophobic groups on the phenyl ring lead to great improvement of the PPARγ binding activity,but introducing multisubstitutions to the phenyl ring could not significantly improve the PPARγ binding activity.
Lastly,we continued to replace the phenyl ring with the heterocyclic aromatic rings and other conjugate groups. As illustrated in Table 2,the replacement of phenyl ring with naphthalene derivatives had no obvious improvements in activity, except for the 4-CH3O substituted naphthalene (2c) which exhibited a preferable PPARγ binding activity (Ki= 45.34 nmol/L). Then,furan ring/thiophene ring was introduced to replace the phenyl ring and simple investigation was undertaken. For compounds 2f,2g,2h,we found that the replacement of furan ring did not result in a big improvement and introducing phenyl ring derivatives to the furan could not promote the binding activity either. 3.3. Binding modes analysis
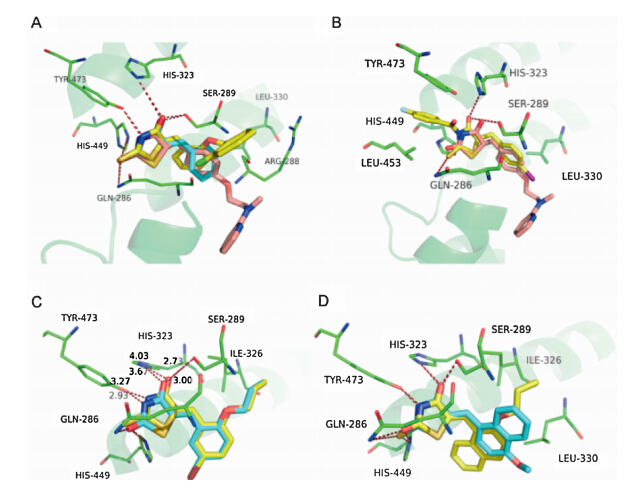
To elucidate the SAR discussed above,we performed molecular fitting of these compounds onto the approved drug Rosiglitazone in the PPARγ ligand binding domain. As illustrated by Fig. 1,it is found that the 2-thioxothiazolidin-4-one scaffold of these compounds fitted well with the TZD group of the Rosiglitazone, and also could form key hydrogen bond interactions with residues His 449,Tyr 473,His 323,Ser 289 and Gln 286 like the Rosiglitazone [5,41]. Contrarily,these compounds exhibited different hydrophobic contacts by substituent groups orienting to other parts of the hydrophobic LBD. For example,compounds 1 and7could make hydrophobic interactions with Arg 288 and Leu 330,respectively (Fig. 2A),which were both far from the Rosiglitazone. Moreover,substituent groups in different positions on the phenyl would lead to different binding conformations in which substituent groups at ortho-positions (compound 7) lie in the upper cave of the LBD while they at meta-positions (compound 1) stretched to Leu 330 with potential hazard of sterical clash. Therefore,larger substituent groups at ortho-positions led to superior binding activities (compounds 5,6,7,10,18 and 19 versus compounds 2 and 4),whilst they at meta-positions would result in an ill effect to binding activities (compounds 8,9,23,24,26 and 28). In addition,although compound 13 could not form the H-bond with Tyr 473 owing to the acetylation of the N atom in the 2-thioxothiazolidin-4-one scaffold,the extending benzyl could makep-pstack interactions with Tyr 473 and VDW interactions with Leu 453 as compensations (Fig. 2B). While turning to the 2-thiocarbonyl of the scaffold,it was observed that all the H-bonds aforementioned might be more stable with corresponding distanced shortened that is driven by the 2-thioxothiazolidin-4-one scaffold of 18 than the traditional TZD scaffold of 29,which led to a 30 folds discrepancy in binding activities at last (Fig. 2C). When the substituent benzyl groups were replaced by similar aromatic rings such as naphthyls,furans and thienyls,it was found that the aromatic rings could also contact to the receptor by VDW interactions with Leu 330 (Fig. 2D),but further derivative groups would bring potential sterical clash in the narrow binding pocket to attenuate the binding activities of these compounds (2c and 2j versus 2a,2b and 2k).

| Download: |
| Fig. 2. Potential binding modes of representative compounds 1,7,13,20,34,2a and 2c in the LBD of PPARγ . All the compounds were superposed onto the Rosiglitazone (in pink) by SHAFTS,the protein is shown in green with key residues labeled out, and potential H-bonds are highlighted with red dashed lines. (A) Compounds 1 and 7 are colored in light blue and yellow,respectively. (B) Compound 13 is shown in yellow. (C) Compounds 20 and 34 are colored in yellow and light blue,respectively, and the distances of potential H-bonds are measured for comparison. (D) Compounds 2a and 2c are shown in yellow and light blue,respectively. | |
{kind=link}
The unsaturated 2-thioxo-4-thiazolidinone derivatives were designed,synthesized,and evaluated for their PPARγ binding activity. The PPARγ competitive binding assays revealed that these derivatives have potent PPARγ binding activities,especially compounds 18 and 38,which exhibited the most potent activities withKi values of 12.15 nmol/L and 14.46 nmol/L,respectively. The structure-activity relationship (SAR) was analyzed to conclude which substitutions were preferred to the series. It was found that the novel scaffold of a 2-thioxo-4-thiazolidinone and an unsaturated linker could retain the PPARγ binding activity,while substitutions of hydrophobic groups to the phenyl ring especially at theortho-ormeta-position would be conducive to the binding activities. Furthermore,through the molecular fitting,we speculated potential receptor-ligand interactions and found new binding modes of these compounds different from Rosiglitazone. These results would inspire us to do further evaluations of these compounds on PPARγ corresponded physiological functions and provide useful information for potential structure-based screening in the future.
Acknowledgment
This work was supported by the National Natural Science Foundation of China (Nos. 81072627,81230090 and 81222046), Shanghai Committee of Science and Technology (Nos. 12431900901 and 12401900801) and the Fundamental Research Funds for the Central Universities (111 Project,No. B07023);
Appendix A. Supplementary data
Supplementary data associated with this article can be found,in the online version,at http://dx.doi.org/10.1016/j.cclet.2014.10.008.
| [1] | M. Ahmadian, J.M. Suh, N. Hah, et al., PPAR [gamma] signaling and metabolism: the good, the bad and the future, Nat. Med. 99 (2013) 557-566. |
| [2] | R.M. Evans, G.D. Barish, Y.X. Wang, PPARs and the complex journey to obesity, Nat. Med. 10 (2004) 355-361. |
| [3] | B.M. Forman, P. Tontonoz, J. Chen, et al., 15-Deoxy-δ 12,14-prostaglandin J2 is a ligand for the adipocyte determination factor PPARgamma, Cell 83 (1995) 803-812. |
| [4] | P. Tontonoz, B.M. Spiegelman, Fat and beyond: the diverse biology of PPARgamma, Annu. Rev. Biochem. 77 (2008) 289-312. |
| [5] | T.M. Willson, P.J. Brown, D.D. Sternbach, B.R. Henke, The PPARs: from orphan receptors to drug discovery, J. Med. Chem. 43 (2000) 527-550. |
| [6] | B. Desvergne, W. Wahli, Peroxisome proliferator-activated receptors: nuclear control of metabolism, Endocr. Rev. 20 (1999) 649-688. |
| [7] | I. Issemann, S. Green, Activation of a member of the steroid hormone receptor superfamily by peroxisome proliferators, Nature 347 (1990) 645-650. |
| [8] | M. Ricote, A.C. Li, T.M. Willson, C.J. Kelly, C.K. Glass, The peroxisome proliferatoractivated receptor-gamma is a negative regulator of macrophage activation, Nature 391 (1998) 79-82. |
| [9] | K. Schoonjans, B. Staels, J. Auwerx, Role of the peroxisome proliferator-activated receptor (PPAR) in mediating the effects of fibrates and fatty acids on gene expression, J. Lipid Res. 37 (1996) 907-925. |
| [10] | S.M. Rangwala, M.A. Lazar, Peroxisome proliferator-activated receptor gamma in diabetes and metabolism, Trends Pharmacol. Sci. 25 (2004) 331-336. |
| [11] | G. Chinetti, J.C. Fruchart, B. Staels, Peroxisome proliferator-activated receptors (PPARs): nuclear receptors at the crossroads between lipid metabolism and inflammation, Inflamm. Res. 49 (2000) 497-505. |
| [12] | P. Delerive, J.C. Fruchart, B. Staels, Peroxisome proliferator-activated receptors in inflammation control, J. Endocrinol. 169 (2001) 453-459. |
| [13] | D.J. Mangelsdorf, C. Thummel, M. Beato, et al., The nuclear receptor superfamily: the second decade, Cell 83 (1995) 835-839. |
| [14] | R.K. Chaturvedi, M.F. Beal, PPAR: a therapeutic target in Parkinson's disease, J. Neurochem. 106 (2008) 506-518. |
| [15] | S.H. Caldwell, E.E. Hespenheide, J.A. Redick, et al., A pilot study of a thiazolidinedione, troglitazone, in nonalcoholic steatohepatitis, Am. J. Gastroenterol. 96 (2001) 519-525. |
| [16] | M. Diamant, R.J. Heine, Thiazolidinediones in type 2 diabetes mellitus, Drugs 63 (2003) 1373-1406. |
| [17] | J.M. Lehmann, L.B. Moore, T.A. Smith-Oliver, et al., An antidiabetic thiazolidinedione is a high affinity ligand for peroxisome proliferator-activated receptor gamma (PPARgamma), J. Biol. Chem. 270 (1995) 12953-12956. |
| [18] | H. Yki-Järvinen, Thiazolidinediones, N. Engl. J. Med. 351 (2004) 1106-1118. |
| [19] | B. Staels, J.C. Fruchart, Therapeutic roles of peroxisome proliferator-activated receptor agonists, Diabetes 54 (2005) 2460-2470. |
| [20] | T.M. Willson, J.E. Cobb, D.J. Cowan, et al., The structure-activity relationship between peroxisome proliferator-activated receptor gamma agonism and the antihyperglycemic activity of thiazolidinediones, J. Med. Chem. 39 (1996) 665-668. |
| [21] | B.A. Neuschwander-Tetri, W.L. Isley, J.C. Oki, et al., Troglitazone-induced hepatic failure leading to liver transplantation: a case report, Ann. Intern. Med. 129 (1998) 38-41. |
| [22] | J. Cohen, Risks of troglitazone apparent before approval in USA, Diabetologia 49 (2006) 1454-1455. |
| [23] | R.A. Daynes, D.C. Jones, Emerging roles of PPARs in inflammation and immunity, Nat. Rev. Immunol. 2 (2002) 748-759. |
| [24] | F. Calon, Y.H. Zhao, C. Julien, J.W. Winkler, et al., Docosahexaenoic acid-derived neuroprotectin D1 induces neuronal survival via secretase- and PPARgamma mediated mechanisms in Alzheimer's disease models, Plos One 6 (2011) 1-15. |
| [25] | H.L.X. Zhang, M. Wei, C. Qin, et al., Neuroprotective effects of pioglitazone in a rat model of permanent focal cerebral ischemia are associated with peroxisome proliferator-activated receptor gamma-mediated suppression of nuclear factorkappaB signaling pathway, Neuroscience 176 (2011) 381-395. |
| [26] | A.L.F. Gabriel Pascual, S. Ogawa, A. Gamliel, et al., A SUMOylation-dependent pathway mediates transrepression of inflammatory response genes by PPARgamma, Nature 437 (2005) 759-763. |
| [27] | M.T. Heneka, T. Klockgether, D.L. Feinstein, Peroxisome proliferator-activated receptor-g ligands reduce neuronal inducible nitric oxide synthase expression and cell death in vivo, J. Neurosci. 20 (2000) 6862-6867. |
| [28] | G. Landreth, Q. Jiang, S. Mandrekar, M. Heneka, PPARgamma agonists as therapeutics for the treatment of Alzheimer's disease, Neurotherapeutics 5 (2008) 481-489. |
| [29] | S.E. Nissen, K. Wolski, Effect of rosiglitazone on the risk of myocardial infarction and death from cardiovascular causes, N. Engl. J. Med. 356 (2007) 2457-2471. |
| [30] | B.M. Psaty, C.D. Furberg, The record on rosiglitazone and the risk of myocardial infarction, N. Engl. J. Med. 357 (2007) 67-69. |
| [31] | L. Azoulay, H. Yin, K.B. Filion, et al., The use of pioglitazone and the risk of bladder cancer in people with type 2 diabetes: nested case-control study, Br. Med. J. 344 (2012) e3645. |
| [32] | J.D. Lewis, A. Ferrara, T. Peng, et al., Risk of bladder cancer among diabetic patients treated with pioglitazone interim report of a longitudinal cohort study, Diabetes Care 34 (2011) 916-922. |
| [33] | A.M. Lincoff, K. Wolski, S.J. Nicholls, S.E. Nissen, Pioglitazone and risk of cardiovascular events in patients with type 2 diabetes mellitus, J. Am. Med. Assoc. 298 (2007) 1180-1188. |
| [34] | C. Day, Thiazolidinediones: a new class of antidiabetic drugs, Diabetic Med. 16 (1999) 179-192. |
| [35] | F.J. Schopfer, M.P. Cole, A.L. Groeger, et al., Covalent peroxisome proliferatoractivated receptor gamma adduction by nitro-fatty acids selective ligand activity and anti-diabetic signaling actions, J. Biol. Chem. 285 (2010) 12321-12333. |
| [36] | C. Yung-Chi, W.H. Prusoff, Relationship between the inhibition constant (KI) and the concentration of inhibitor which causes 50 per cent inhibition (I50) of an enzymatic reaction, Biochem. Pharmacol. 22 (1973) 3099-3108. |
| [37] | X. Liu, H. Jiang, H. Li, SHAFTS: a hybrid approach for 3D molecular similarity calculation. 1. Method and assessment of virtual screening, J. Chem. Inf. Model. 51 (2011) 2372-2385. |
| [38] | X. Liu, F. Bai, S. Ouyang, et al., Cyndi: a multi-objective evolution algorithm based method for bioactive molecular conformational generation, BMC Bioinformatics 10 (2009) 101. |
| [39] | R.T. Nolte, G.B. Wisely, S. Westin, et al., Ligand binding and co-activator assembly of the peroxisome proliferator-activated receptor-gamma, Nature 395 (1998) 137-143. |
| [40] | G. Bruno, L. Costantino, C. Curinga, et al., Synthesis and aldose reductase inhibitory activity of 5-arylidene-2,4-thiazolidinediones, Bioorg. Med. Chem. 10 (2002) 1077-1084. |
| [41] | A.G. Chittiboyina, M.S. Venkatraman, C.S. Mizuno, et al., Design and synthesis of the first generation of dithiolane thiazolidinedione- and phenylacetic acid-based PPARgamma agonists, J. Med. Chem. 49 (2006) 4072-4084. |