




b Department of Polymer Science and Engineering, University of Science and Technology Beijing, Beijing 100083, China
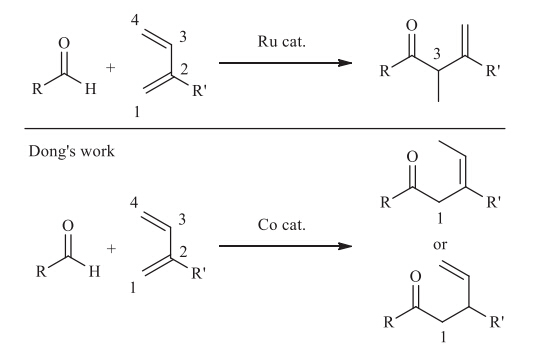
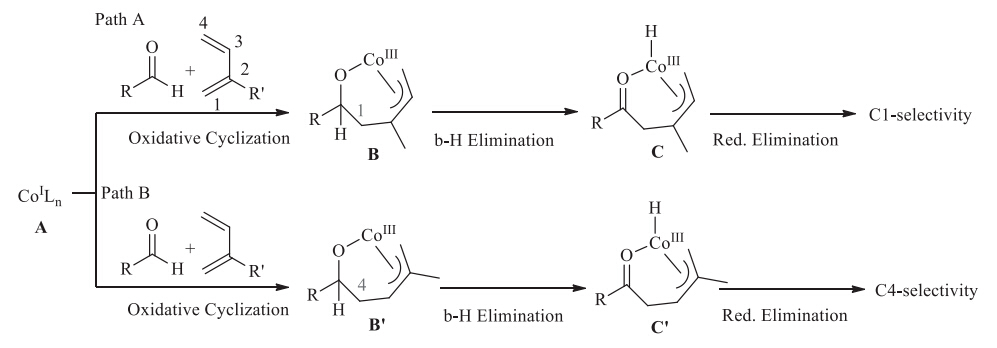
First-row transition metals are more abundant and less expensive than precious transition metals like Pd,Rh and Ru,and this character promotes the rapid development of various first-row metals catalyzed cross-coupling reactions [1]. In this context,Cocatalyzed organic transformations such as cycloaddition reactions, reduction reactions,Aldol reactions,Michael reactions,hydrovinylation reactions,hydroacylation reactions have achieved many attractive results [2]. Another amazing reason for the exploitation of first-row transition metals is that a different chemical- or regioselectivity could be realized by replacing the precious transition metal catalyst with the first-row transition metal catalyst. For example,in the Ru-catalyzed hydroacylation of 2-substituted 1,3-diene reported by Krische [3] and Ryu [4],C-C bond selectively forms at the C3 site (Scheme 1). However,Dong et al.recently reported Co-catalyzed hydroacylation of 2-substituted 1,3-diene,in which C-C bond selectively forms at the C1 site [5]. As to the mechanism of Co-catalyzed hydroacylation of 1,3-diene,Donget al. proposed the oxidative cyclization pathway (Path A,Scheme 2). In this mechanism,the pre-catalyst CoII complex is first reduced by In/InBr3to the active CoI complex A[5,6,7]. Then C1-selective oxidative cyclization occurs on CoI complex to give the CoIII complex B. β-H elimination occurs on B give the hydride complex C. Finally,C-H reductive elimination on C gives the product and regenerate the catalyst A. Similar to Path A,the oxidative cyclization of 1,3-diene and aldehyde also possibly occurs at the C4 site,which will finally produce a C4-selective product (Path B,Scheme 2). Interestingly,only trace amount of C4-selective products were obtained in Dong’s reactions.

| Download: |
| Scheme 1.Different regioselectivity of hydroacylation of 1,3-diene by different transition-metal catalysts. | |
{kind=link}

| Download: |
| Scheme 2.Proposed mechanism for C1-selectivity in Co-catalyzed hydroacylation of 1,3-diene and its competitive mechanism for C4-selectivity. | |
{kind=link}
Our recent research interests cover the Co-catalyzed crosscouplings,such as the investigations on origin for ligand controlled regioselectivity in Co-catalyzed hydroarylation of styrene [8]. In the present study,we systematically investigated the mechanism of Co-catalyzed hydroacylation of 1,3-diene with DFT method. The calculation results indicate that β-H elimination is the ratedetermining step in the proposed catalytic cycle. C4-selective oxidative cyclization is kinetically disfavored than the pathway of C1-selective oxidative cyclization and its following steps. Further analysis shows that both the electronic and steric effects of the 2-substituted group of 1,3-diene contribute to the dominance of C1-selective oxidative cyclization. 2. Computational details
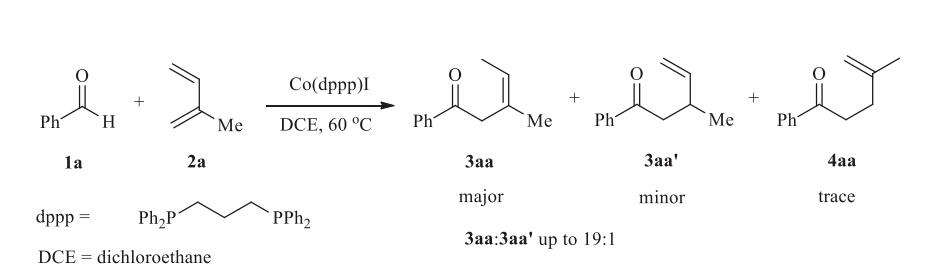
Co-catalyzed cross coupling of benzaldehyde 1a and 2-methyl-1,3-butene 2a with the dppp ligand (dppp = 1,3-bis(diphenylphosphino)propane) was chosen as the model reaction for mechanistic studies (Scheme 3). Computational studies were performed with Gaussian 09 program [9,10,11,12,13,14,15,16,17,18,19,20,21,22,23]. Geometry optimization was conducted in gas phase with M06 method [24]. The effective core potential of LANL2DZ with the associated valence basis set [25] was used describe Co and I atoms while 6-31G(d) basis set was used for the other atoms. At the same level of theory,frequency analysis was performed to confirm that the optimized structure was either a minimum or a transition state,and also to obtain the thermodynamic energy correction. To reduce the overestimated entropy effect due to the gas-phase optimizations,Sakaki’s strategy by omitting the electronic and rotational entropic contributions is used [26,27]. In addition,solution-phase single point energies were calculated with a larger basis set based on the optimized structure,i.e. SDD [28] for Co and I while 6-311+G(d,p) for the rest atoms. SMD model was used for solution-phase single point energy calculations (solvent = dichloroethane) [29]. The reported energies were the solution-phase single point energies corrected by gas-phase Gibbs free energy corrections,corresponding to 1 mol/L and 298 K.

| Download: |
| Scheme 3.Model reaction for mechanistic study. | |
{kind=link}
The energy profile of Path A was investigated first and the results were shown in Fig. 1. The ligand exchange of the neutral CoI complex 1 with aldehyde 1a and diene 2a gives the cationic CoI complex 2,which causes an energy decrease of 22.8 kcal/mol. In complex 2,Co is coordinated by the C3 and C4 double bond (Fig. 1). Then the oxidative cyclization occurs viathe transition state TS1 and generates the intermediate 3. This step further causes an energy decrease of 8.5 kcal/mol,and its energy barrier is only 4.5 kcal/mol. With the formation of the new C-C bond, the lengths of several other C-C bonds of 2a are changed during the transformation from 2 to 3. The C1-C2 bond is lengthened from 1.342 Å to 1.491 Å,C2-C3 bond is shortened from 1.472 Å to 1.386 Å,C3-C4 bond is lengthened from 1.389 Å to 1.428 Å. Meanwhile,the C-O bond of1achanged from 1.235 Å to 1.393 Å. Besides,the coordination mode of 2a changes from η2 (in 2)to η3 (in 3).

| Download: |
| Fig. 1. Energy profile of Path A and the C4-selective oxidative cyclization step. | |
{kind=link}
The intermediate 3 then undergoes β-H elimination viathe four-membered transition state TS2 to generate the complex 4a.In this step,a remarkable distortion of the cyclometallic ring occurs to make the hydrogen atom on the phenyl-substituted carbon get close to the Co center. As a result,a high energy barrier forb-H elimination (20.8 kcal/mol from 3 to TS2 ) is required. Along with the transformation from 3 to 4a,the C-O bond of 1a shortens to 1.241 Å,which is close to the C=O bond length of 1a in complex 2.In 4a,Co is coordinated by a carbonyl group and an allyl anion. The carbonyl locatescisto the hydride and the allyl anion locates trans to the hydride.
From the intermediate 4a,the C-H reductive elimination could occur directlyviatransition state TS3a,and generates the product 3aa '. The free energy of TS3a is +13.2 kcal/mol. Note that C-H elimination is also possibly occurs on the isomers of 4a,i.e. 4b and 4c. The carbonyl group locatescisto the hydride in 4b while locates transto the hydride in 4c. C-H reductive elimination occurs on 4b viatransition state TS3b to generate the product 3aa ,and the free energy of TS3b is +8.4 kcal/mol. From4c,both reductive transition states TS3c and TS3d are possible because C4 and C2 both locatecis to the hydride. 3aa and 3aa'are formed via TS3c and TS3d , respectively. The free energies of TS3c and TS3d are +16.2, +13.7 kcal/mol. Among the four reductive elimination transition states,TS3b is the most stable one. As the result,the formation of 3aa is kinetically favored. Note that the experimental yields of different products (i.e. 3aa > 3aa') were well reproduced by our calculations,while the calculation results tend to slightly overestimate the energy difference between TS3a and TS3b .
After C-H reductive elimination via TS3b ,the complex 5 is formed. The ligand exchange of 5 with 2-methyl-1,3-diene and benzaldehyde finally affords the product 3aa and also regenerates the reactive catalyst 2 to finish the whole catalytic cycle. The free energy change of whole reaction is -7.2 kcal/mol and b-H elimination is the rate-determining step.
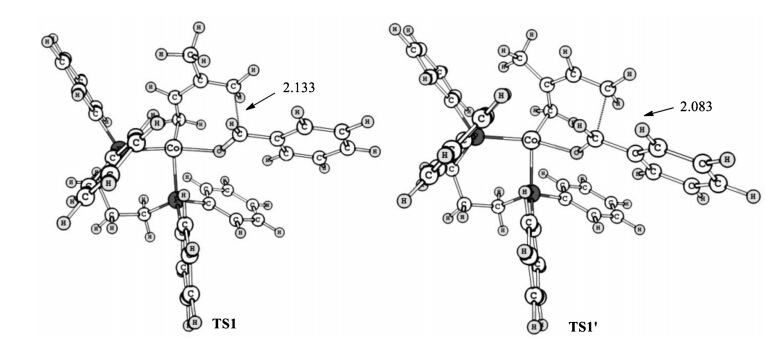
We next examined the possibility of Path B in which oxidative cyclization occurs at the C4 site of 2a and finally leads to the C4-selective product 4aa. The related oxidative cyclization transition state is TS1' in which Co is coordinated by the C1 and C2 site of 2a in an η2 fashion. The free energy of TS1' is +24.4 kcal/mol,higher than that of TS1 by +11.4 kcal/mol,indicating that TS1' is kinetically disfavored. TS1' is less stable than all of the species in the C1-selective pathway,indicating that C1-selective oxidative cyclization and the following steps are more favored. Therefore C1-selective product 3aa is the predicted product of this mechanism whereas 4aa should be trace. On this basis,we explore the reason for the C1-selectivity and investigated the electronic distribution of 2a. It is found that due to the electrondonating methyl group attached on C2 site,the Mulliken charge on the C1 to C4 atoms of 2a are -0.262,+0.330,+0.179 and -0.065, respectively. In this case,the electrophilic attack of carbonyl carbon atom of 1aon the more electron-rich C1 atom will be favored than that on C4. We further compared the structures of TS1 and TS1' (Fig. 2). It is found that the methyl group on C2 site locates more close to the ligand in TS1',thus a more significant repulsion is expected in TS1'. In one word,TS1' is a less favored than TS1 due to both the electronic and steric effects of the methyl group on C2 site.

| Download: |
| Fig. 2. The optimized structures of oxidative cyclization transition states. Bond length are given in A. | |
{kind=link}
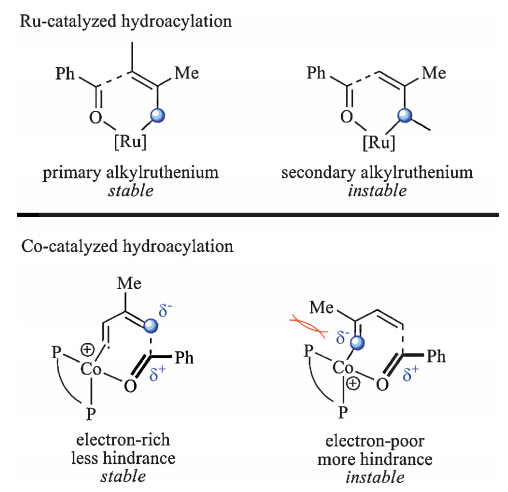
In the present study,we also made efforts in distinguishing the differences between the Ru- and Co-catalyzed hydroacylation of 1,3-diene. Ru-catalyzed hydroacylation of 2a generates C3-selective products [3,4]. The proposed mechanism includes hydrometalation of 1,3-diene,carbonyl addition and beta-H elimination steps. This catalytic cycle has been recently investigated by Meng et al.,implying that carbonyl addition is the rateand regioselectivity-determining step [30]. The hydrometallation of Ru-H complexes on 1,3-diene mainly generates the primary alkylruthenium intermediate because of the steric effect of the substitute on 1,3-diene. The following carbonyl additionviaa six membered transition state leads to C3-selective product. Different from the Ru-catalyzed hydroacylation mechanism,Co-catalyzed hydroacylation start from oxidative cyclization. In this step,the coordination site of Co on 1,3-diene is not restricted to C1 and C3 but to C1 and C4. Because C1 and C4 both become primary carbon anions after oxidative cyclization,and their steric effect becomes quite similar. On the other hand,in the seven membered transition states of oxidative cyclization,electronic effect of the substitute on 1,3-diene and the steric repulsion between it and the ligand both prefer C1-selectivity. As a result,Co-catalyzed hydroacylation of 1,3-diene generates C1-selective product (Scheme 4).

| Download: |
| Scheme 4.Comparison of the regioselectivity in Ru- and Co-catalyzed hydroacylation of 1,3-diene. | |
{kind=link}
Co-catalyzed hydroacylation of 1,3-diene provides powerful synthetic tools for the access to unsaturated carbonyl compounds, and its unique regioselectivity makes this method complementary to the related Ru-catalyzed reactions. DFT methods were used to study the mechanism of Co-catalyzed hydroacylation of 2-methyl-1,3-butene. Computational results indicate that the catalytic cycle includes oxidative cyclization,β-H elimination and C-H reductive elimination steps. The C1-hydroacylation is dominant compared with the C4-hydroacylation pathway,because the activation barrier for the first step (i.e. oxidative cyclization) in the C4-pathway is even higher than the overall activation barrier of C1-pathway.β-H elimination is the rate-determining step in the whole catalytic cycle of C1-pathyway because of the significant distortion of cyclometalic ring. The methyl substituent on C2 site makes C1 more electronic rich than C4,thus promotes the electrophilic attack of carbonyl carbon of aldehydes on C1. Besides, the methyl group locates closer to the ligand in C4-selective oxidative cyclization transition state,which also contributes the difficulty of the C4-pathway. Therefore,both the electronic and steric effects favor the C1-hydroacylation mechanism.
Acknowledgments
We thank the NSFC (Nos. 21325208,21172209,21361140372, 21202006),SRFDP (No. 20123402110051),FRFCU (No. WK2060190025),CAS (No. KJCX2-EW-J02),Fok Ying Tung Education Foundation,Anhui Provincial Natural Science Foundation (No. 1308085QB38),China National Grid Project funded by MOE of China and the supercomputer center of Shanghai and USTC.
Appendix A. Supplementary data
Supplementary data associated with this article can be found,in the online version,at http://dx.doi.org/10.1016/j.cclet.2014.10.021.
| [1] | C. Wang, Y. Fu, L. Liu, Q.X. Guo, New advance of Fe- and Co-catalyzed C-C coupling reactions, Chin. J. Org. Chem. 27 (2007) 703-723. |
| [2] | H. Pellissier, H. Clavier, Enantioselective cobalt-catalyzed transformations, Chem. Rev. 114 (2014) 2775-2823. |
| [3] | F. Shibahara, J.F. Bower, M.J. Krische, Diene hydroacylation from the alcohol or aldehyde oxidation level via ruthenium-catalyzed C-C bond-forming transfer hydrogenation: synthesis of β,γ-unsaturated ketones, J. Am. Chem. Soc. 130 (2008) 14120-14122. |
| [4] | S. Omura, T. Fukuyama, J. Horiguchi, Y. Murakami, I. Ryu, Ruthenium hydridecatalyzed addition of aldehydes to dienes leading to β,γ-unsaturated ketones, J. Am. Chem. Soc. 130 (2008) 14094-14095. |
| [5] | Q.A. Chen, D.K. Kim, V.M. Dong, Regioselective hydroacylation of 1,3-dienes by cobalt catalysis, J. Am. Chem. Soc. 136 (2014) 3772-3775. |
| [6] | M.A. Bohn, A. Schmidt, G. Hilt, M. Dindaroğlu, H. Schmalz, Cobalt-catalyzed 1,4- hydrobutadienylation of 1-Aryl-1,3-dienes with 2,3-dimethyl-1,3-butadiene, Angew. Chem. Int. Ed. 50 (2011) 9689-9693. |
| [7] | L. Fiebig, J. Kuttner, G. Hilt, et al., Cobalt catalysis in the gas phase: experimental characterization of cobalt(I) complexes as intermediates in regioselective Diels- Alder reactions, J. Org. Chem. 78 (2013) 10485-10493. |
| [8] | Z.W. Yang, H.Z. Yu, Y. Fu, Mechanistic study on ligand-controlled cobalt-catalyzed regioselectivity switchable hydroarylation of styrenes, Chem. Eur. J. 19 (2013) 12093-12103. |
| [9] | M.J. Frisch, G.W. Trucks, H.B. Schlegel, et al., Gaussian 09 Revision D.01, Wallingford, CT, 2013. |
| [10] | X.J.Du, Y.H. Tang, X. Zhang, M. Lei,A theoretical study on the alkene insertion step in Rh-Yanphos catalyzed hydroformylation, Chin. Chem. Lett. 24 (2013) 1083-1086. |
| [11] | T.C. Jiang, Z.Y. Wang, B.B. Du, S.S. Zhao, Theoretical characterization of hole mobility in BTBPD, Chin. Chem. Lett. 24 (2013) 945-948. |
| [12] | T.J. Gong, Y.Y. Jiang, Y. Fu, Rh(I)-catalyzed borylation of primary alkyl chlorides, Chin. Chem. Lett. 25 (2014) 397-400. |
| [13] | Q. Zhou, Y. Li, 1,3-Cationic alkylidene migration of nonclassical carbocation: a density functional theory study on gold(I)-catalyzed cycloisomerization of 1,5-enynes containing cyclopropene moiety, J. Am. Chem. Soc. 136 (2014) 1505-1513. |
| [14] | B. Lu, Y. Li, Y. Wang, et al., [3,3]-Sigmatropic rearrangement versus carbene formation in gold-catalyzed transformations of alkynyl aryl sulfoxides: mechanistic studies and expanded reaction scope, J. Am. Chem. Soc. 135 (2013) 8512-8524. |
| [15] | R. Shang, Z.W. Yang, Y. Wang, S.L. Zhang, L. Liu, Palladium-catalyzed decarboxylative couplings of 2-(2-azaaryl)acetates with aryl halides and triflates, J. Am. Chem. Soc. 132 (2010) 14391-14393. |
| [16] | Y.F. Yang, G.J. Cheng, P. Liu, et al., Palladium-catalyzed meta-selective C-H bond activation with a nitrile-containing template: computational study on mechanism and origins of selectivity, J. Am. Soc. Chem. 136 (2014) 344-355. |
| [17] | S. Zhang, L. Shi, Y. Ding, Theoretical analysis of the mechanism of palladium(Ⅱ) acetate-catalyzed oxidative Heck coupling of electron-deficient arenes with alkenes: effects of the pyridine-type ancillary ligand and origins of the metaregioselectivity, J. Am. Chem. Soc. 133 (2011) 20218-20229. |
| [18] | L. Li, F. Wu, S. Zhang, et al., A heteroleptic cyclometalated iridium(Ⅲ) fluorophenylpyridine complex from partial defluorohydrogenation reaction: synthesis, photophysical properties and mechanistic insights, Dalton Trans. 42 (2013) 4539-4543. |
| [19] | S. Qu, Y. Dang, C. Song, et al., Catalytic mechanisms of direct pyrrole synthesis via dehydrogenative coupling mediated by PNP-Ir or PNN-Ru pincer complexes: crucial role of proton-transfer shuttles in the PNP-Ir System, J. Am. Chem. Soc. 136 (2014) 4974-4991. |
| [20] | Y. Dang, S. Qu, Z.X. Wang, X. Wang, A computational mechanistic study of an unprecedented Heck-type relay reaction: insight into the origins of regio- and enantioselectivities, J. Am. Chem. Soc. 136 (2014) 986-998. |
| [21] | Z. Dong, C.H. Liu, Y. Wang, M. Lin, Z.X. Yu, Gold(I)-catalyzed endo-selective intramolecular a-alkenylation of b-yne-furans: synthesis of seven-memberedring- fused furans and DFT calculations, Angew. Chem. Int. Ed. 52 (2013) 14157-14161. |
| [22] | Y. Fu, Z. Li, S. Liang, Q.X. Guo, L. Liu, Mechanism for carbon-oxygen bond-forming reductive elimination from palladium(IV) complexes, Organometallics 27 (2008) 3736-3742. |
| [23] | H.Z. Yu, Y.Y. Jiang, Y. Fu, L. Liu, Alternative mechanistic explanation for liganddependent selectivities in copper-catalyzed N- and O-arylation reactions, J. Am. Chem. Soc. 132 (2010) 18078-18091. |
| [24] | Y. Zhao, D.G. Truhlar, The M06 suite of density functionals for main group thermochemistry, thermochemical kinetics, noncovalent interactions, excited states, and transition elements: two new functionals and systematic testing of four M06-class functionals and 12 other functionals, Theor. Chem. Acc. 120 (2008) 215-241. |
| [25] | P.J. Hay, W.R. Wadt, Ab initio effective core potentials for molecular calculations - potentials for the transition-metal atoms Sc to Hg, J. Chem. Phys. 82 (1985) 270-283. |
| [26] | Z. Li, S.L. Zhang, Y. Fu, Q.X. Guo, L. Liu, Mechanism of Ni-catalyzed selective C-O bond activation in cross-coupling of aryl esters, J. Am. Chem. Soc. 131 (2009) 8815-8823. |
| [27] | Z. Li, Y. Fu, S.L. Zhang, Q.X. Guo, L. Liu, Heck-type reactions of imine derivatives: a DFT study, Chem. Asian J. 5 (2010) 1475-1486. |
| [28] | Y.Y. Jiang, Y. Fu, L. Liu, Mechanism of palladium-catalyzed decarboxylative crosscoupling between cyanoacetate salts and aryl halides, Sci. China Chem. 55 (2012) 2057-2062. |
| [29] | A.V. Marenich, C.J. Cramer, D.G. Truhlar, Universal solvation model based on solute electron density and a continuum model of the solvent defined by the bulk dielectric constant and atomic surface tensions, J. Phys. Chem. B 113 (2009) 6378-6396. |
| [30] | Q. Meng, F. Wang, M. Li, Ruthenium hydride-catalyzed regioselective addition of benzaldehyde to dienes leading to β,γ-unsaturated ketones: a DFT study, J. Mol. Model 18 (2012) 4955-4963. |