




As we know,the 1,2,3-triazole ring is not only a hydrogen-bond donor but also a linking unit in the structure scaffold. Its planar structure may facilitate π stacking interaction with target enzymes similar to phenyl rings. The 1,2,3-triazole based derivatives possess a wide variety of biological properties,for example, antiviral [1],agonist [2],antibacterial [3],anti-HIV [4, 5],and so on. Presently,the most popular method for the construction of the 1,2,3-triazole framework is the 1,3-dipolar cycloaddition reaction of azides with alkynes [6, 7, 8],which is called the click reaction. Click reactions are modular,tolerant of a wide range of solvents and functional groups,simple to perform,and very high yielding. However,most of these reactions can be processed by using copper (I) as the catalyst. Recently,a simple method for the preparation of the 1,2,3-triazole moiety from benzyl azide and active methylene compounds under mild conditions using inorganic base has been reported [9, 10].
In the past three decades,calixarenes have been targets of basic and applied sciences. Specifically,calixarenes have become a popular building block for the preparation of new host systems in supramolecular chemistry [11],because calixarenes are easily amenable to the upper or lower rim functionalization of the macrocyclic skeleton. To the best of our knowledge,copper (I)- assisted 1,3-dipolar azide-alkyne cycloaddition (CuAAC) is the main synthetic approach for introduction of 1,2,3-triazole moiety into the calixarene scaffold both at their lower and upper rim [12]. Therefore,developing metal-free and more practical and efficient synthetic approaches for calixarene derivatives bearing the 1,2,3- triazole unit is desirable. In this paper,we report the preparation of novel calix[4]arene derivatives containing the 1,2,3-triazole moiety from calix[4]arene-based azide and acetyl acetone or ethyl acetoacetate by using 1,3-dipolar cycloaddition reaction under anhydrous potassium carbonate as catalyst. 2. Experimental
Unless otherwise noted,all materials were obtained from commercial suppliers and dried and purified by standard procedures. The melting point was measured on an SGW X-4 monocular microscope melting point apparatus with an unadjusted thermometer. 1H NMR and 13C NMR spectra were acquired on a 400 MHz Bruker Avance spectrometer with CDCl3 as the solvent and tetramethylsilane (TMS) as the internal standard. The chemical shifts were reported in δ (ppm). Mass spectra (MS) data were obtained using an Esquire 6000 Mass Spectrometer. Thermogravimetry (TG) was recorded on a Perkin-Elmer thermal analyzer at a heating rate of 10 ℃ min-1. Column chromatography was performed with silica gel (200-300 mesh,Qingdao Haiyang Chemical Co.,Ltd.,China).
Depicted in Scheme 1 is our synthetic route developed to allow for efficient introduction of the 1,2,3-triazole moiety on the calix[4]arene skeleton toward the end of the synthesis. The synthesis of 4 and 5 starts with treatment of tert-butylcalix[4]- arene 1 [13] with 1,2-dibromoethane in CH3CN to give compound 2 [14]. Replacement of the bromine of 2 with sodium azide was carried out in DMF to obtain intermediate 3 [15]. Then, calix[4]arene-based azide 3 underwent 1,3-dipolar cycloaddition reaction with acetyl acetone or ethyl acetoacetate in the base medium (anhydrous potassium carbonate) to afford the title compounds 4 and 5 with good yields (81% and 63%,respectively).

|
Download:
|
| Scheme 1.Synthesis route of calix[4]arene derivatives 4 and 5. Reagents and conditions: (i) K2CO3/CH3CN,reflux,24 h,yield 87%; (ii) NaN3/DMF,40-50 ℃,5 h,yield 85%; (iii) K2CO3/DMF,70 ℃,6-12 h,yield 81% for 4 and 63% for 5. | |
General procedure for the synthesis of calix[4]arene derivatives 4 and 5: The calix[4]arene-based azide 3 (0.8 g,1 mmol),acetyl acetone (0.4 g,4 mmol) or ethyl acetoacetate (0.52 g,4 mmol), anhydrous potassium carbonate (0.83 g,6 mmol),and DMF (40 mL) were added to a round bottom flask equipped with a stirrer. The reaction mixture was agitated at 70 ℃ for 6-12 h. The progress of the reaction was monitored by TLC. After the completion of the reaction,the solvent was removed under vacuum. The residual mass was quenched in the ice-water mixture and neutralized with 5% HCl solution. The product was extracted with dichloromethane dried over anhydrous sodium sulfate. Evaporation of the solvent yielded the crude product,which was purified by flash chromatography on silica gel eluted with petroleum ether/ethyl acetate (8:1-6:1). 3. Results and discussion
The structures of the compounds synthesized herein were fully confirmed by MP,1H NMR,13C NMR,and mass spectra (ESI-MS). In the 1H NMR spectra of compounds 4 and 5,two singlets for the tert-butyl groups (absorptions of the doublets near δ 0.8 and 1.3),two doublets for the bridging methylene groups (absorptions of the doublets near δ 3.4 and 4.0,J = 13.2 Hz for ArCH2Ar protons),and two doublets for the aromatic protons (absorptions of the doublets near δ 6.6 and 7) suggested that 4 and 5 were in a cone conformation. The chemical shift values are close to those reported for other calix[4]arene molecules in the same conformations [16, 17]. In ESI mass spectra of the two compounds,both 4 and 5 showed [M-H]-peaks. The characterization data of the targeted compounds are listed below.
Compound 4: White solid,yield: 81%; mp: 268-270 ℃; 1H NMR (400 MHz,CDCl3) δ 0.82 (s,18H,C(CH3)3),1.30 (s,18H, C(CH3)3),2.56 (s,6H,COCH3),2.84 (s,6H,CH3-triazole),3.26 (δ,4H, J = 13.2 Hz,ArCH2Ar),3.89 (δ,4H,J = 13.2 Hz,ArCH2Ar),4.39 (t,4H, J = 6.0 Hz,OCH2CH2-triazole),4.92 (t,4H,J = 6.0 Hz,OCH2CH2-triazole),6.60 (s,4H,ArH),7.03 (s,4H,ArH),8.23 (s,2H,ArOH); 13C NMR (100 MHz,CDCl3) δ 194.4,150.1,149.1,147.5,143.6,141.9,138.7,131.6,127.5,125.6,125.1,73.4,46.8,33.9,33.8,31.7,30.9,30.8,27.6,9.5; MS (ESI): m/z 950.3 [M-H]-.
Compound 5: White solid,yield: 63%; mp: 216-218 ℃; 1H NMR (400 MHz,CDCl3) δ 0.84 (s,18H,C(CH3)3),1.28 (s,18H, C(CH3)3),1.40 (t,6H,J = 7.2 Hz,OCH2CH3),2.88 (s,6H,COCH3),3.40 (δ,4H,J = 13.2 Hz,ArCH2Ar),4.15 (d,4H,J = 13.2 Hz,ArCH2Ar),4.42 (q,4H,J = 7.2 Hz,OCH2CH3),4.70 (t,4H,J = 5.2 Hz,OCH2CH2-triazole),4.95 (t,4H,J = 5.2 Hz,OCH2CH2-triazole),6.65 (s,4H,ArH),7.02 (s,4H,ArH),8.86 (s,2H,ArOH); 13C NMR (100 MHz,CDCl3) δ 168.5,152.1,149.7,146.6,143.7,142.0,134.5,133.4, 127.0,125.9,125.0,74.2,60.7,46.6,39.9,33.7,31.8,30.8,30.6, 14.2,9.6; MS (ESI): m/z 1010.2 [M-H]-.
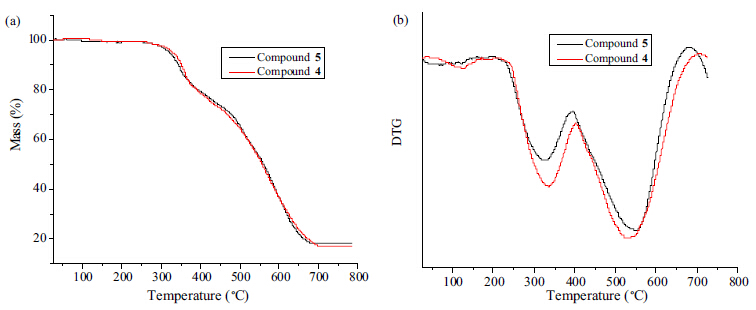
The thermal stability of 4 and 5 was examined by the thermogravimetric analyses at a heating rate of 10 ℃ min-1 from ambient temperature to 800 ℃ in flowing nitrogen atmosphere. The thermoanalytical results obtained from TG and DTG curves of the compounds 4 and 5 were summarized in Table 1 and the TG and DTG curves were illustrated in Fig. 1.
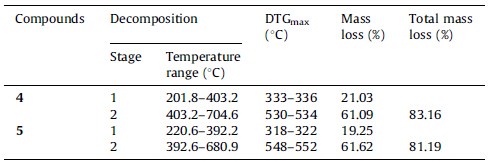
| Table 1 Thermoanalytical results of the compounds 4 and 5. |
{kind=link}

|
Download:
|
| Fig. 1.TG (a) and DTG (b) curves of the compounds 4 and 5. | |
{kind=link}
The TG and DTG curves clearly show that the mass losses of the compounds 4 and 5 containing 1,2,3-triazole groups are similar to each other. The mass loss of the compound 4 takes place slowly, whereas the first and second stages mass loss of the compound become sharp as shown by the DTGmax at 333-336 ℃ and 530- 534 ℃. In the first and second stages,the thermal mass losses of the synthesized compound 4 occur between 201.8 and 403.2 ℃ and completed in the interval 403.2-704.6 ℃ together with the mass losses between 21.03% and 61.09%. Similarly,it is seen that the mass loss of the compound 5 in the first and second stages appear at 220.6-392.2 ℃ and 392.6-680.9 ℃,respectively; the mass losses of this compound were determined as 19.25% and 61.62%, respectively. Obviously,the second thermal mass loss stage of the two title compounds is the basic decomposition stage and the decomposition mechanism of the compounds 4 and 5 are complex. 4. Conclusion
We have applied 1,3-dipolar cycloaddition reaction using anhydrous potassium carbonate as catalyst to the synthesis of calix[4]- arene derivatives containing 1,2,3-triazole moiety. The metal-free simple synthetic procedures and good yields should make this type of reaction a general method to construct complicated macrocyclic skeletons. Thermal analysis shows that the basic mass loss stage of the title compounds containing 1,2,3-triazole groups clearly took place between 392 ℃ and 403 ℃. The further synthesis and functional study for similar molecules are underway in our laboratory. Acknowledgments The authors gratefully thank the financial supports of the National Natural Science Foundation of China (No. 21102003), Scientific Research Foundation for the Introduction of Talent and Young Teachers Scientific Research Foundation of Anhui University of Science & Technology (Nos. 11214,2012QNY27).
| [1] | X.M. Chen, Z.J. Li, Z.X. Ren, Z.T. Huang, Synthesis of glucosylated 1,2,3-triazole derivatives, Carbohydr. Res. 315 (1999) 262-267. |
| [2] | L.L. Brockunier, E.R. Parmee, H.O. Ok, et al., Human beta3-adrenergic receptor agonists containing 1,2,3-triazole-substituted benzenesulfonamides, Bioorg. Med. Chem. Lett. 10 (2000) 2111-2114. |
| [3] | M.J. Genin, D.A. Allwine, D.J. Anderson, et al., Substituent effects on the antibacterial activity of nitrogen-carbon-linked (azolylphenyl)oxazolidinones with expanded activity against the fastidious gram-negative organisms Haemophilus influenza and Moraxella catarrhalis, J. Med. Chem. 43 (2000) 953-970. |
| [4] | R. Alvarez, S. Velazquez, A. San-Felix, et al., 1,2,3-Triazole-20,50-bis-O-(tert-butyl-dimethylsilyl)-b-D-ribofuranosyll-3'-spiro-5"-(4'-amino-l",2"-oxathiole 2",2"-dioxide) (TSAO) analogues: synthesis and anti-HIV-1 activity, J. Med. Chem. 37 (1994) 4185-4194. |
| [5] | S. Velazquez, R. Alvarez, C. Perez, et al., Regiospecific synthesis and anti-human immuno-deficiency virus activity of novel 5-substituted N-alkylcarbamoyl and N, N-dialkyl carbamoyl 1,2,3-triazole-TSAO analogues, J. Antiviral Chem. Chemother. 9 (1998) 481-489. |
| [6] | R. Huisgen, in: A. Padwa (Ed.), 1,3-Dipolar Cycloaddition Chemistry, vol. 1, Wiley, New York, 1984, pp. 1-176. |
| [7] | F. Xie, K. Sivakumar, Q. Zeng, et al., A fluorogenic ‘click' reaction of azidoanthracene derivatives, Tetrahedron 64 (2008) 2906-2914. |
| [8] | K. Sivakumar, F. Xie, B. Cash, et al., A fluorogenic 1,3-dipolar cycloaddition reaction of 3-azidocoumarins and acetylenes, Org. Lett. 6 (2004) 4603-4606. |
| [9] | V.R. Kamalraj, S. Senthil, P. Kannan, One-pot synthesis and the fluorescent behavior of 4-acetyl-5-methyl-1,2,3-triazole regioisomers, J. Mol. Struct. 892 (2008) 210-215. |
| [10] | I.F. Cottrell, D. Hands, P.G. Houghton, G.R. Humphrey, S.H.B. Wright, An improved procedure for the preparation of 1-benzyl-1H-1,2,3-triazoles form benzyl azides, J. Heterocycl. Chem. 28 (1991) 301-304. |
| [11] | (a) Y. Liu, C. You, H. Zhang, Supramolecular Chemistry-Molecular Recognition and Assembly of Synthetic Receptors, Nankai University Press, 2001; (b) L. Mandolini, R. Ungaro, Calixarenes in Action, Imperial College Press, London, 2000; (c) Z. Asfari, W. Bohmer, J. Harrowfield, et al., Calixarenes, Kluwer Academic Press, Dordrecht, 2001. |
| [12] |
(a) A. Vecchi, B. Melai, A. Marra, C. Chiappe, A. Dondoni, Microwave-enhanced ionothermal CuAAC for the synthesis of glycoclusters on a calix[4]arene platform, J. Org. Chem. 73 (2008) 6437-6440; (b) S.Y. Park, J.H. Yoon, C.S. Hong, et al., A pyrenyl-appended triazole-based calix[4]arene as a fluorescent sensor for Cd2+ and Zn2+, J. Org. Chem. 73 (2008) 8212-8218; (c) B.T. Zhao, X.M. Zhu, X.H. Chen, Z.N. Yan, W.M. Zhu, Novel clicked tetrathiafulvalene-calix[4]arene assemblies: synthesis and intermolecular electron transfer toward p-chloranil, Chin. Chem. Lett. 24 (2013) 573-577; (d) H. Chen, Z.L. Zou, S.L. Tan, et al., Efficient synthesis of water-soluble calix[4]-arenes via thiol-ene "click" chemistry, Chin. Chem. Lett. 24 (2013) 367-369. |
| [13] | C.D. Gutsche, M. Iqbal, D. Stewart, Calixarenes. 19. Syntheses procedures for ptertbutylcalix[4]arene, J. Org. Chem. 51 (1986) 742-745. |
| [14] | Z.T. Li, G.Z. Ji, C.X. Zhao, et al., Self-assembling calix[4]arene catenanes preorganization, conformation, selectivity, and efficiency, J. Org. Chem. 64 (1999) 572-3584. |
| [15] | F. Szemes, D. Hesek, Z. Chen, et al., Synthesis and characterization of novel acyclic, macrocyclic, and calix[4]arene ruthenium(Ⅱ) bipyridyl receptor molecules that recognize and sense anions, Inorg. Chem. 35 (1996) 5868-5879. |
| [16] | V.S. Talanov, R.A. Bartsch, Highly selective preparation of conformationally rigidstereoisomeric calix[4]arenes with two carboxymethoxy groups, J. Chem. Soc., Perkin Trans. 1 (1999) 1957-1961. |
| [17] | C. Jaime, J. de Mendoza, P. Prados, P. Nieto, C. Sanchez, Carbon-13 NMR chemical shifts. A single rule to determine the conformation of calix[4]arenas, J. Org. Chem. 56 (1991) 3372-3376. |