




N-9 substituted purine nucleosides have long been recognized as core structures in biological systems [1, 2] and are of great pharmaceutical importance,such as antivirus drugs,Aciclovir and Famciclovir [3, 4, 5, 6]. Traditionally,the alkylation of purines with halogenated compounds [7],mesylate [8],or tosylate [9] is an important synthetic route to purine nucleoside derivatives, which usually suffers from poor regioselectivity,resulting in a mixture of N-9 and N-7 isomers. The challenging issue of regioselectivity between two competitive tautomeric forms (N-7-H and N-9-H) makes the development of effective methods for synthesis of N-9 substituted purine derivatives highly desirable [10].
Recently,the addition reaction involves purine rings has been made much progress for its regioselectivity. Lira and Huffman [11] and Wang et al. [12] reported the base-catalyzed Michael addition of purines with α.β-unsaturated compounds. Peel et al. [13] demonstrated the Pd-catalyzed addition reaction of purines with cyclopentadiene monoepoxide to give the N-9 carbocyclic nucleosides exclusively. Moreover,the addition reactions between purines and 3,4-dihydropyran,got high regioselectivity, while it suffered from unsatisfactory yield for extensive use because the product could be decomposed under the acidic conditions [14, 15]. Therefore,the further study on the addition reaction involves purine rings is still of great significance for the development of novel and direct access to N-9 substituted purine nucleosides.
Ammonium triflates have emerged as a kind of highly efficient and cost-effective Lewis acid catalyst in various chemical transformations [16, 17, 18, 19]. They have many advantages including ease of product separation,recycling of the catalyst and environmental acceptability as compared to traditionally Lewis acid catalysts. In continuation of our interest in the application of novel ammonium triflate organocatalysts [20, 21],herein,we report an efficient and environmentally friendly method for the synthesis of N-9-alkoxyalkylated purines via highly regioselective N-alkoxyalkylation of purines with vinyl ethers catalyzed by Lproline triflate (L-ProT) (Scheme 1).

|
Download:
|
| Scheme 1.Selective N-alkoxyalkylation of purine rings. | |
Analytical grade solvents and commercially available reagents were used without further purification. The flash column chromatography was carried out over silica gel (200-400 mesh), purchased from Qingdao Haiyang Chemical Co.,Ltd. Melting points were determined on a Bu¨chi B-540 capillary melting point apparatus and uncorrected. 1H NMR and 13C NMR spectra were recorded at VARAIN-400 using DMSO-d6 or CDCl3 as the solvent with tetramethylsilane (TMS) as an internal standard. Chemical shifts are given in δ relative to TMS,the coupling constants J are given in Hz. Mass spectra were measured with Thermo Finnigan LCQ-Advantage. HRMS analyze were measured on an Agilent 6210 TOF LC/MS using ESI or EI (electrospray ionization) techniques.
General procedures for the synthesis of N-9 substituted purine nucleoside derivatives: A mixture of purine (1 mmol),vinyl ether (2 mmol),and L-ProT (0.05 mmol) was stirred in AcOEt (2 mL) at room temperature. After completion of the reaction (monitored by TLC),the mixture was added AcOEt or CH2Cl2 (20 mL) and then washed with water (10 mL × 2). The organic layer was dried over anhydrous Na2SO4,filtered and evaporated. Purification by column chromatography on silica gave the product. Physical and chemical data of the chosen product is demonstrated below.
2,6-Dichloro-9-(tetrahydro-2H-pyran-2-yl)-9H-purine (3a): White powder; 93% yield; mp 115-117 ℃; 1H NMR (400 MHz, CDCl3): δ 8.36 (s,1H),5.77 (dd,1H,J = 10.4,2.4 Hz),4.23-4.14 (m, 1H),3.79 (td,1H,J = 11.6,2.8 Hz),2.22-1.68 (m,6H); 13C NMR (100 MHz,CDCl3): δ 152.8,152.1,151.5,143.7,130.7,82.6,69.0, 32.1,24.9,22.7. HRMS-ESI: calcd. for C10H10C12N4NaO: 295.0124; found: 295.0130. 3. Results and discussion
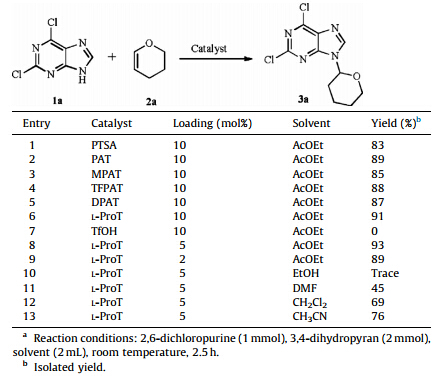
We started our investigation by choosing 2,6-dichloropurine and 3,4-dihydropyran as model substrates for the optimization of reaction conditions. Ammonium triflate catalysts were easily to be removed by washing with water after the completion of reaction and proved more efficient than the traditional catalyst p-toluenesulfonic acid (PTSA) (Table 1,entries 1-6). From Table 1,L-ProT proved to be the best catalyst with a 91% yield of 3a,while others,including aniline triflate (PAT),N-methylaniline triflate (MPAT),trifluoromethyl aniline triflate (TFPAT), diphenylammonium triflate (DPAT),gave lower yields of 85%- 89% (Table 1,entries 2-6). Product 3a was not observed when using trifluoromethanesulfonic acid (TfOH) as the catalyst (Table 1,entry 7). And a sufficient amount of 5 mol% L-ProT was necessary for this reaction (Table 1,entries 8 and 9). Then the solvents were examined and the results were summarized in Table 1. Only trace amount of product was obtained in EtOH (Table 1,entry 10). And higher yields were given in DMF,CH2Cl2, and CH3CN,but results were still unsatisfactory (Table 1,entries 11-13). So AcOEt was chosen as the optimum solvent for the reaction. Moreover,the further screening of reaction temperature and time confirmed that reaction could be finished at room temperature within 2.5 h. It should be noted that there was no N-7 isomer [22] or other byproduct in above reactions. The product 3a was examined as a racemic mixture,which indicated that the catalysis did not influence the enantioselectivity of the reaction.
| Table 1 The optimization of reaction conditions. a |
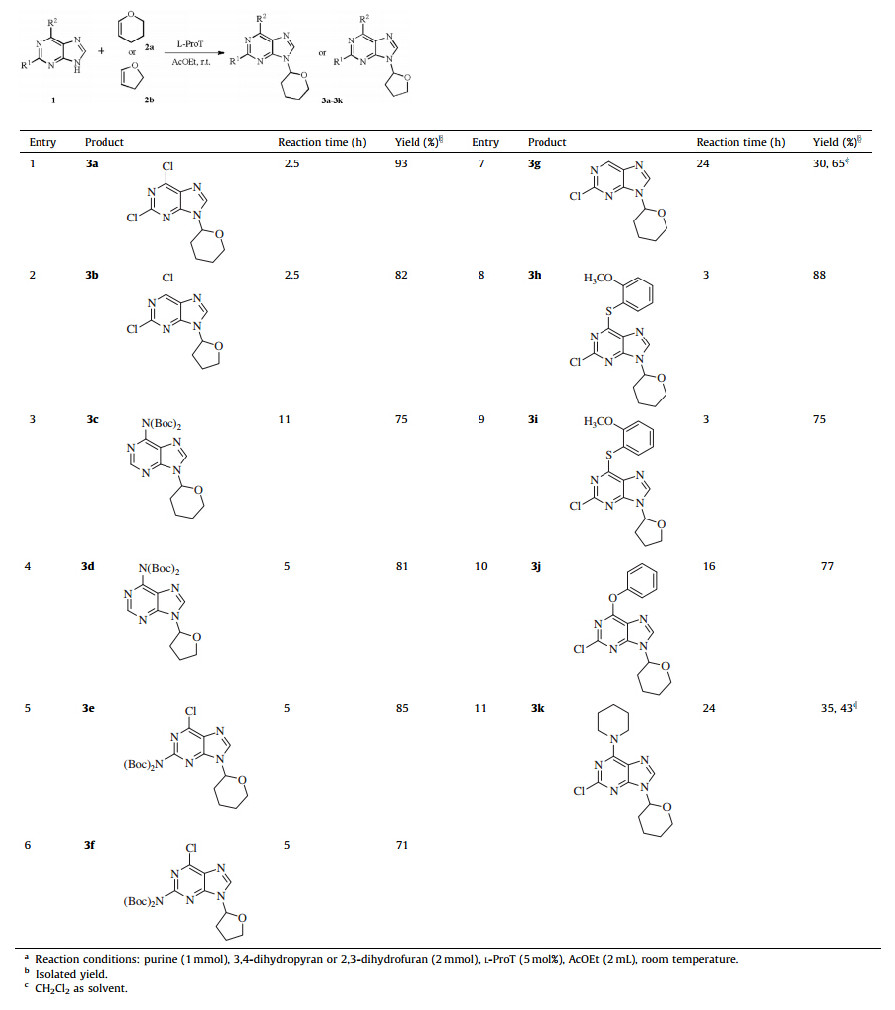
Under the optimized reaction conditions,various substituted purines were subjected to the reaction as shown in Table 2. The Boc-protected adenine,Boc-protected 6-chloroguanine and chlorinated purines reacted smoothly with 3,4-dihydropyran or 2,3-dihydrofuran in good yields (Table 2,entries 1-7). A variety of groups,such as Cl and Boc were well tolerated,allowing the further conversion of these nucleoside analogues. The purines with different substituents at the C-6 position,including a-methoxy phenyl thiol,phenoxy,piperidyl also gave expected products in moderate to good yields (Table 2,entries 8-11). 2-Chloro-purine and 2-chloro-6-(piperidin-1-yl)-purine,which have poor solubility in AcOEt,were conducted in CH2Cl2with moderate yields (Table 2, entries 7 and 11). From Table 2,the circinal vinyl ethers,including 3,4-dihydropyran and 2,3-dihydrofuran,were proved to be reliable reaction substrates.
| Table 2 The N-alkoxyalkylation of various purines. a |
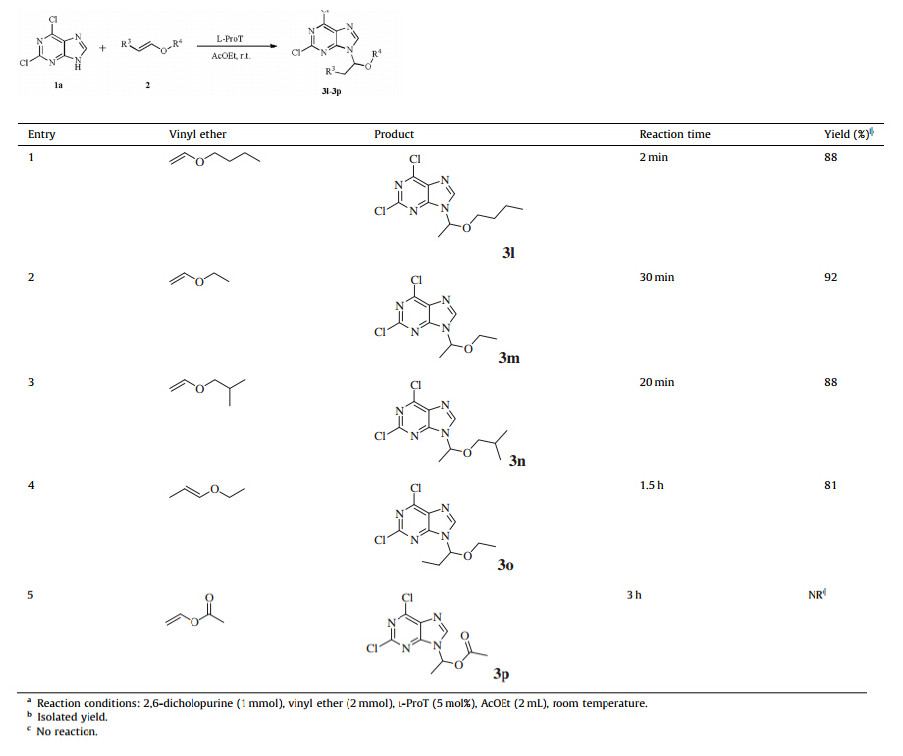
After that,various open chain vinyl ethers were also examined to evaluate the general applicability of the method. The chain vinyl ethers with different alkyl groups afforded the corresponding products 3l-3n in excellent yields of 88%-92% (Table 3,entries 1- 3). And the mixtures of cis- and trans-ethyl propenyl ethers were also proved to be reliable reaction substrates,further illustrating that steric hindrance of the structure has little influence on the reaction except reaction rate (Table 3,entry 4). However,vinyl acetate could not give the desired product 3p because of the electron-withdrawing effect of acetoxy group on the vinyl group (Table 3,entry 5). Besides,the reaction of 2,6-dichloropurine and D-(+)-glucal was conducted under the optimized reaction conditions,we surprisingly found that compound 3q was produced in a 53% yield (Scheme 2).
| Table 3 The reaction of 2,6-dicholopurine with various vinyl ethers. a |
{kind=link}

|
Download:
|
| Scheme 2.Reaction of 2,6-dichloropurine and D-(+)-glucal. | |
{kind=link}
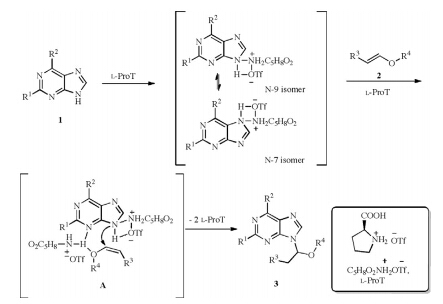
A plausible mechanism for the N-alkoxyalkylation of various purines in the presence of L-ProT is proposed in Scheme 3. Purine firstly reacted with L-ProT to form intermediates of N-9 isomer and N-7 isomer [21]. The complex of N-9 isomer further reacted with vinyl ether at the N-3 position [23] in the presence of L-ProT to form a complex (A),followed by nucleophilic addition reaction to afford the thermodynamically stable N-9 alkoxyalkylated purines exclusively [15].

|
Download:
|
| Scheme 3.Proposed mechanism for the reaction of purines and vinyl ethers. | |
{kind=link}
In summary,we have developed an alternative route to N-9-alkoxyalkylated nucleobases through highly regioselective alkoxyalkylation of purines with vinyl ethers catalyzed by L-ProT. It is characterized by high regioselectivity,employment of costeffective catalyst,mild conditions. Besides,the L-ProT catalyzed addition reaction shows general tolerance to a variety of functionalities,such as Cl and Boc,making this method quite attractive. Further application of this methodology to synthesize more promising N-9-alkoxyalkylated purine nucleosides is now underway in our laboratory.
Acknowledgments We are grateful to the Natural Science Foundation of Zhejiang Province (No. LY13B020015) and Zhejiang Key Innovation Team of Green Pharmaceutical Technology (No. 2012R10043-07) for financial support.| [1] | H. Rosemeyer, The chemodiversity of purine as a constituent of natural products, Chem. Biodivers 1 (2004) 361-401. |
| [2] | H.B. Xu, R.F. Ye, S.Y. Yang, R. Li, X. Yang, Electrochemical DNA nano-biosensor for the detection of genotoxins in water samples, Chin. Chem. Lett. 25 (2014) 29-34. |
| [3] | E.D. Clercq, Antiviral drugs: current state of the art, J. Clin. Virol. 22 (2001) 73-89. |
| [4] | P. Lu, S.H. Jiang, J.X. Liu, Y.S. She, R.Y. Ji, Design, synthesis and anti-HBV activity of novel bis(trifluoroethyl)phosphonomethyl ether derivatives of acyclovir, Chin. Chem. Lett. 20 (2009) 507-510. |
| [5] | W.B. Parker, Enzymology of purine and pyrimidine antimetabolites used in the treatment of cancer, Chem. Rev. 109 (2009) 2880-2893. |
| [6] | E. de Clercq, Ten paths to the discovery of antivirally active nucleoside and nucleotide analogues, Nucleosides Nucleotides Nucleic Acids 31 (2012) 339-352. |
| [7] | U. Diederichsen, D. Weicherding, N. Diezemann, Side chain homologation of alanyl peptide nucleic acids: pairing selectivity and stacking, Org. Biomol. Chem. 3 (2005) 1058-1066. |
| [8] | M. Kitamatsu, A. Takahashi, T. Ohtsuki, M. Sisido, Synthesis of pyrrolidine-based oxy-peptide nucleic acids carrying four types of nucleobases and their transport into cytoplasm, Tetrahedron 66 (2010) 9659-9666. |
| [9] | N.F. Zakirova, A.V. Shipitsyn, E.F. Belanov, M.V. Jasko, A new approach to the synthesis of optically active alkylated adenine derivatives, Bioorg. Med. Chem. Lett. 14 (2004) 3357-3360. |
| [10] | M. Gandelman, E.N. Jacobsen, Highly enantioselective catalytic conjugate addition of N-heterocycles to α,β-unsaturated ketones and imides, Angew. Chem. Int. Ed. 44 (2005) 2393-2397. |
| [11] | E.P. Lira, C.W. Huffman, Some Michael-type reactions with adenine, J. Org. Chem. 31 (1965) 2188-2191. |
| [12] | H. Wu, Z.Q. Tian, L.L. Zhang, Y.D. Huang, Y.M. Wang, Organocatalytic enantioselective aza-Michael addition of purine bases to α,β-unsaturated ketones, Adv. Synth. Catal. 354 (2012) 2977-2984. |
| [13] | M.R. Peel, D.D. Sternbach, M.R. Johnson, A short, enantioselective synthesis of the carbocyclic nucleoside carbovir, J. Org. Chem. 56 (1991) 4990-4993. |
| [14] | T. Borrmann, A. Abdelrahman, R. Volpini, et al., Structure-activity relationships of adenine and deazaadenine derivatives as ligands for adenine receptors, a new purinergic receptor family, J. Med. Chem. 52 (2009) 5974-5989. |
| [15] | D. Singh, M.J. Wani, A. Kumar, A simple solution to the age old problem of regioselective functionalization of guanine: first practical synthesis of acyclic N9-and/or N7-guanine nucleosides starting from N2,N9-diacetylguanine, J. Org. Chem. 64 (1999) 4665-4668. |
| [16] | T. Funatomi, K. Wakasugi, T. Misaki, Y. Tanabe, Pentafluorophenylammonium triflate (PFPAT): an efficient, practical, and cost-effective catalyst for esterification, thioesterification, transesterification, and macrolactone formation, Green Chem. 8 (2006) 1022-1027. |
| [17] | B. Karimi, M. Ghoreishi-Nezhad, Highly chemoselective acetalization of carbonyl compounds catalyzed by a novel recyclable ammonium triflate-functionalized silica, J. Mol. Catal. A: Chem. 277 (2007) 262-265. |
| [18] | L. Mercs, G. Pozzi, S. Quici, Efficient condensation of carboxylic acids with alcohols catalyzed by fluorous ammonium triflates, Tetrahedron Lett. 48 (2007) 3053-3056. |
| [19] | N. Montazeri, S. Mahjoob, Highly efficient and easy synthesis of 2,4,6-triarylpyr-idines catalyzed by pentafluorophenylammonium triflate (PFPAT) as a new recyclable solid acid catalyst in solvent-free conditions, Chin. Chem. Lett. 23 (2012) 419-422. |
| [20] | J.J. Li, L.M. Lu, W.K. Su, A new strategy for the synthesis of benzoxanthenes catalyzed by proline triflate in water, Tetrahedron Lett. 51 (2010) 2434-2437. |
| [21] | J.J. Li, P. He, C.M. Yu, DPAT-catalyzed one-pot regioselective synthesis of polysubstituted pyridines and 1,4-dihydropyridines, Tetrahedron 68 (2012) 4138-4144. |
| [22] | K. Groebke, J. Hunziker, W. Fraser, et al., Chemistry of a-amino nitriles. Part 9. Why pentose-and not hexose-nucleic acids? Part 5. Purine-purine pairing in homo-DNA. Guanine, isoguanine, 2,6-diaminopurine, and xanthine, Helv. Chim. Acta 81 (1998) 375-474. |
| [23] | M. Miyaki, B. Shimizu, N→N Alkyl and glycosyl migration of purines and pyrimidines. II. Glycosyl migration of 3-glycosyl-N6-acyladenine derivatives, Chem. Pharm. Bull. 18 (1970) 732-740. |