




b Chongqing Key Laboratory of Chemical Process for Clean Energy and Resource Utilization, Chongqing University, Chongqing 400044, China;
c College of Materials Science and Engineering, Chongqing University of Technology, Chongqing 400054, China
Imidoesters have been widely used in the synthesis of nitrogencontaining organic compounds,and frequently used as cross-linking reagents as well as drugs participation in the human body reactions [1, 2, 3, 4, 5, 6]. In 1883,Pinner reported the first synthesis of imidoesters with nitrile and alcohol in the presence of dry hydrochloride [7]. Various kinds of methods have been developed in synthesizing imidoesters since Pinner’s seminal work,using cyanates [8, 9, 10], aldehydes [11],N-hydroxypyridine-2-thioketone sodium salt [12], and aniline [13] as starting materials. However,the synthetic of structurally diverse imidoesters need to be developed regardless of these tremendous efforts devoted to this area.
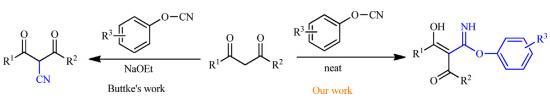
In Buttke’s report,the treatment of dicarbonyl compounds and cyanatobenzenes produces 2-cyano-1,3-diketones under ice-bath conditions in the presence of sodium ethoxide [14]. When we carried out these reactions in the absence of additives,imidoesters were obtained instead of 2-cyano-1,3-diketones (Scheme 1). In our former work,we have reported on theoretical cyanation of aldimine,Cs2CO3-catalyzed transesterification,primary amine-catalyzed aldol condensation,synthesis of bis(indolyl)-methanes,theoretical halo-exchange SN2 reactions,iodination of sp3 C-H with hypervalent iodide,Fe(III)-promoted oxidative free radical coupling of anilines,and superacid promoted benzylation of arenes [15]. Herein we are pleased to report a new method for neat atom-economical synthesis of imidoesters employing cyanatobenzenes and dicarbonyl compounds as starting materials under additive-free conditions.

|
Download:
|
| Scheme 1.Reported cyanation and proposed imidoesterification of dicarbonyl compounds with cyanatobenzenes. | |
Ethyl acetoacetate 2a (126 mL,1.0 mmol) was added to the solution of cyanatobenzene 1a (130 mL,1.2 mmol) in diethyl ether (2 mL). The reaction mixture was stirred at room temperature for 40 h in the atmosphere and monitored by TLC. After completion of the reaction,the volatile components were removed using a vacuum rotary evaporator. The resulting residue was purified by column chromatography on silica gel column using EtOAc/ petroleum ether (1:20 to 1:4,v/v) as eluent to afford the desired product ethyl 3-hydroxy-2-(imino(phenoxy)methyl)but-2-enoate 3a. Products were recorded by NMR spectra on a 500 spectrometer (500 MHz for 1H,125 MHz for 13 C) with deuterated chloroform (CDCl3) as a solvent at 20-25 ℃. For detailed experimental procedure,characterization data of the products,and copies of 1H NMR and 13C NMR spectra,see Supporting information. 3. Results and discussion
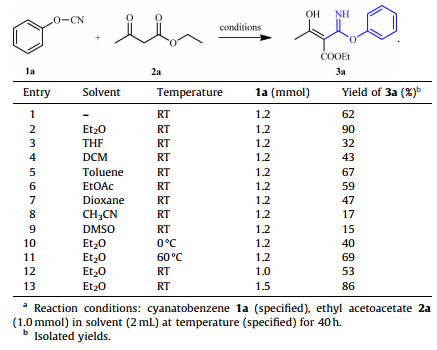
We firstly aimed at the optimization of reaction conditions for the synthesis of imidoesters. The treatment of cyanatobenzene 1a with ethyl acetoacetate 2a was chose as a model reaction to screen various kinds of solvents,temperature and material ratio (Table 1). Under the solvent-free conditions,the imidoester 3a was obtained in 62% yield in the absence of additive (entry 1),while the usage of Et2O as solvent led to better yield of 90% at room temperature (entry 2). Other common solvents such as THF,DCM,toluene, EtOAc,dioxane,acetonitrile and DMSO generally resulted in the decrease of yields to some different extent (entries 3-9). In the examination of temperature,either high or low temperature was found to be unbeneficial to imidoesterification compared with the result obtained at the room temperature (entries 10 and 11). The alternation of the proportion of 1a in the range of 1.0-1.5 equiv. to ethyl acetoacetate 2a was investigated as well. As a result,both less and excessive amount of 2a led to lower yields and 1.2 equiv. of 2a was most appropriate to this transformation (entries 12 and 13). Therefore,the optimal reaction conditions were established: dicarbonyl compounds (1.0 mmol) and cyanatobenzenes (1.2 equiv.) in diethyl ether at room temperature.
| Table 1 Optimization of reaction conditions. a |
{kind=link}
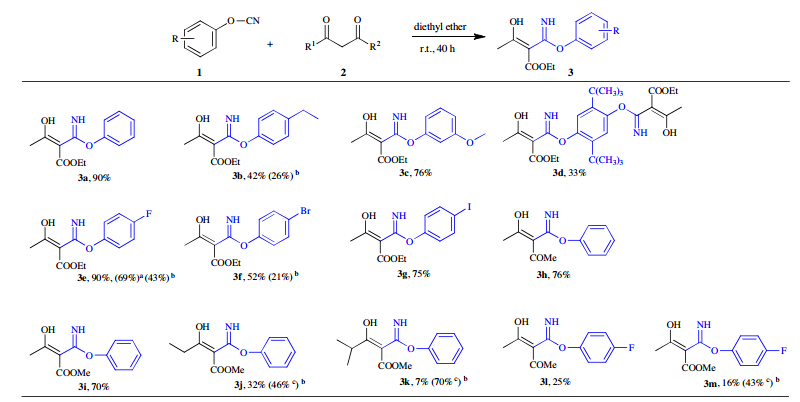
With the established optimal conditions in hand,the scope of cyanatobenzenes was then investigated. Ethyl acetoacetate 2a was used as nucleophile and ethyl 3-hydroxy-2-(imino(phenoxy)methyl)but-2-enoate 3a was obtained providing a high yield up to 90% (Scheme 2). These results showed that the reactivity is independent to electron nature of cyanatobenzenes. With electrodonating substitutions on benzene ring of cyanatobenzenes,ethyl 2-((4-ethylphenoxy)(imino)methyl)-3-hydroxybut-2-enoate 3b was gotten in a yield of 42%,while ethyl 3-hydroxy-2-(imino(3-methoxyphenoxy)methyl)but-2-enoate 3c was produced in a higher yield of 76%. Regarding to the dicyanato material 1,4-di-tert-butyl-2,5-dicyanatobenzene 1d,a low yield of 33% was provided furnishing a diimidoesterified adduct. With electro-withdraw halogenated substitutions on benzene ring of cyanatobenzenes,ethyl 2-((4-fluorophenoxy)(imino)methyl)-3-hydroxybut-2-enoate 3e was obtained in an excellent yield of 90%, whereas ethyl 2-((4-bromophenoxy)(imino)methyl)-3-hydroxybut-2-enoate 3f and ethyl 3-hydroxy-2-(imino(4-iodophenoxy)-methyl)but-2-enoate 3g were obtained in good yields of 52% and 75%,respectively. With an attempt to improve the reactivity through the enamine and enol anion activation (Scheme 3), secondary and tertiary amines such as pyrrole and triethylamine served as additives. As a result,they failed to afford 3b,3e and 3f in comparative yields. In the presence of pyrrole,the decreased conversions of 26%,43% and 21% into 3b,3e and 3f occurred. Likewise,triethylamine had the similar negative effects as well, resulting in a decreased yield of 69% into 3e.

|
Download:
|
| Scheme 2.Expansion of cyanatobenzenes and dicarbonyl compounds. Reaction conditions: dicarbonyl compounds 2 (1.0 mmol),cyanatobenzenes 1 (1.2 equiv.) in diethyl ether (2 mL) at room temperature for 40 h. ( a In the presence of triethylamine (0.2equiv.); b In the presence of pyrrole (0.2 equiv.); c Dicarbonyl compounds 2 (2.0 mmol), cyanatobenzenes 1 (1.2 equiv.) in diethyl ether (2 mL) at room temperature.) | |
{kind=link}

|
Download:
|
| Scheme 3.Two types of activation of dicarbonyl compounds | |
{kind=link}
To further explore the scope of dicarbonyl compounds,a variety of dicarbonyl compounds were then examined. Reacted with electrophilic cyanatobenzene 1a,desired adducts of phenyl 2-acetyl-3-hydroxybut-2-enimidate 3h and methyl 3-hydroxy-2-(imino(phenoxy)methyl)but-2-enoate 3i in 76% and 70% yields were obtained,respectively,whereas lower yields of 32% and 7% were produced for methyl 2-((4-fluorophenoxy)(imino)-methyl)-3-hydroxypent-2-enoate 3j and methyl 3-hydroxy-2-(imino(phenoxy)methyl)-4-methylpent-2-enoate 3k. Whereas 4-fluorophenyl 2-acetyl-3-hydroxybut-2-enimidate 3l and methyl 2-((4-fluorophenoxy)(imino)methyl)-3-hydroxybut-2-enoate 3m were assembled in lower yields of 25% and 16%,respectively. It is worthy noting that in the imidoesterifications of 3j,3k and 3m, the yields of imidoesters were improved to some different extent oppositely in the presence of pyrrole. Based on our understanding of keto-enol tautomerism and nucleophilic addition,a plausible mechanism directed towards the imidoester formation has been proposed (Scheme 4). Through the first tautomerism of 1,3-diketones to active enol A,a nucleophilic addition occurs subsequently to form intermediate B. Then,1,5-H shift from oxygen of carbonyl to imine anion gives rise to the b-keto imidoester C. Finally,the exclusive enol form of imidoester D is generated via 1,3-H shift and the structure of enol is confirmed definitely by 1H NMR analysis.

|
Download:
|
| Scheme 4.A proposed mechanism. | |
{kind=link}
In summary,we have developed a convenient and environmentally benign method for atom-economical synthesis of structurally diverse imidoesters using dicarbonyl compounds and cyanatobenzenes as starting materials. The nucleophilic addition spontaneously can proceed at room temperature in yields up to 90% under additive-free conditions. An acceptable mechanism has been proposed,which is directed towards the imidoester formation.
Acknowledgments We gratefully acknowledge for funding from the SRTP of Chongqing University for undergraduate cultivation (H. Ma and Y. He),the National Natural Science Foundation of China (Nos. 21372265 and 61271059),the Scientific Research Foundation for the Returned Overseas Chinese Scholars,State Education Ministry,the Natural Science Foundation Project of CQ CSTC (Nos. CSTC2013jcyjA0217 and CSTC2010BB4086),and the Fundamental Research Funds for the Central Universities (Nos. CQDXWL-2013-Z012 and CDJZR-14225502). Appendix A. Supplementary data Supplementary data associated with this article can be found,in the online version,at http://dx.doi.org/10.1016/j.cclet.2014.06.008.| [1] | A. Dutton, M. Adams, S.J. Singer, Bifunctional imidoesters as cross-linking reagents, Biochem. Biophys. Res. Commun. 23 (1966) 730-739. |
| [2] | A. Ruoho, P.A. Bartlett, A. Dutton, S.J. Singer, A disulfide-bridge bifunctional imidoester as a reversible cross-linking reagent, Biochem. Biophys. Res. Commun. 63 (1975) 417-423. |
| [3] | E.S. Hand, W.P. Jencks, Mechanism of the reaction of imido esters with amines, J. Am. Chem. Soc. 84 (1962) 3505-3514. |
| [4] | A. Shaw, G.V. Marinetti, The effect of imidoesters, fluorodinitrobenzene and trinitrobenzenesulfonate on ion transport in human erythrocytes, Chem. Phys. Lipids 27 (1980) 329-335. |
| [5] | S. Batmanghelich, R.C. Brown, J.S. Woodhead, I. Weeks, K. Smith, Preparation of a chemiluminescent imidoester for the non-radioactive labelling of proteins, J. Photochem. Photobiol. B 12 (1992) 193-201. |
| [6] | C.A. Presant, S. Parker, Imidoester inhibition of lymphocyte DNA synthesis, Cancer Res. 39 (1979) 345-348. |
| [7] | A. Pinner, Ueber die umwandlung der nitrile in imide, Eur. J. Inorg. Chem. 16 (1883) 1643-1655. |
| [8] | E. Grigat, R. Pütter, Preparation of pyrimidines from cyanic esters and CH-acidic compounds, Angew. Chem. Int. Ed. 4 (1965) 877-878. |
| [9] | D. Martin, H.J. Herrmann, S. Rackow, K. Nadolski, Addition reactions of cyanic esters, Angew. Chem. Int. Ed. 4 (1965) 73-74. |
| [10] | J.M. Fedé, S. Jockusch, N. Lin, R.A. Moss, N.J. Turro, Aryloxy radicals from diaryloxydiazirines: α-cleavage of diaryloxycarbenes or excited diazirines? Org. Lett. 5 (2003) 5027-5030. |
| [11] | P. Yin, W.B. Ma, Y. Chen, et al., Highly efficient cyanoimidation of aldehydes, Org. Lett. 11 (2009) 5482-5485. |
| [12] | J.L. Esker, M. Newcomb, Chemistry of amidyl radicals produced from Nhydroxypyridine-2-thione imidate esters, J. Org. Chem. 58 (1993) 4933-4940. |
| [13] | R.W. Leiby, Synthesis of 3-amino-4(3H)-quinazolinones from N-(2-carbomethoxyphenyl) imidate esters, J. Org. Chem. 50 (1985) 2926-2929. |
| [14] | K. Buttke, H.J. Niclas, A convenient and improved procedure for the cyanation of enamines and 1,3-dicarbonyl compounds, Synth. Commun. 24 (1994) 3241-3248. |
| [15] |
(a) S.Y. Zheng, J.Y. Wang, Y. Xiong, Theoretical investigation on isomerization of Et2AlCN to Et2AlNC and cyanation of aldimine, J. Mol. Struct. Theochem. 869 (2008) 83-88; (b) S.Y. Zheng, Y. Xiong, J.Y. Wang, Theoretical studies on identity SN2 reactions of lithium halide and methyl halide: a microhydration model, J. Mol. Model. 16 (2010) 1931-1937; (c) Y. Xiong, X.Q. Zhang, Significant heterogeneous carbonate salt catalyzed acetylation of alcohols via a transesterification process with carbonate salt activated alcohol 1H NMR evidence, Chin. J. Chem. 29 (2011) 1143-1148; (d) Y. Xiong, S.T. Zhang, X.G. Ling, X. Zhang, J.Y. Wang, Theoretical investiga-tion on identical anionic halide-exchange SN2 reaction processes on N-haloammonium cation NH3X+ (X = F, Cl, Br, and I), Int. J. Quantum Chem. 112 (2012) 2475-2481; (e) X. Zhang, Y. Xiong, S.T. Zhang, et al., Aldol condensations of aldehydes and ketones catalyzed by primary amine on water, Asian J. Chem. 24 (2012) 751-755; (f) X.G. Ling, Y. Xiong, S.T. Zhang, R.F. Huang, X.H. Zhang, Effective synthesis of benzyl halides triggered by in situ prepared hypervalent halides, Chin. Chem. Lett. 24 (2013) 45-48; (g) X.G. Ling, Y. Xiong, R.F. Huang, et al., Synthesis of benzidine derivatives via FeCl3;6H2O-promoted oxidative coupling of anilines, J. Org. Chem. 78 (2013) 5218-5226; (h) X.F. Xu, Y. Xiong, X.G. Ling, et al., A practical synthesis of bis(indolyl)methanes catalyzed by BF3;Et2O, Chin. Chem. Lett. 25 (2014) 406-410; (i) Y. Li, Y. Xiong, X.M. Li, et al., Benzylation of arenes with benzyl ethers promoted by the in situ prepared superacid BF3-H2O, Green Chem. 16 (2014) 2976-2981. |