




L-Sugars often have biological activities of medicinal or agricultural value [1],since they have been found in many bioactive oligosaccharides,antibiotics,glycopeptides,terpene glycosides,as well as steroid glycosides and clinically useful nucleosides. For examples,L-glucose is present in the natural product,(-)-littoralisone,known as a bioactive agent for increasing NGF-induced neurite outgrowth in PC12D cells [2]. L-Gulopyranosides are the key constituents of the antitumor drug,Bleomycin A2 [3] and the nucleoside antibiotic,Adenomycin [4]. The repeating disaccharide units of heparin [5],heparin sulphate, and dermatan sulphate glycosaminoglycans contain L-iduronic acid [6]. Moreover,L-rhamnose and L-mannose are found in the extracellular polysaccharides S-130,S-88,and S-198 [7],which are useful in the industry of food and medicine. The phenolic derivatives of L-mannose that are found in some steroidal glycosides are potent substrates for measuring the a-L-mannosidase activity of commercial naringinase [8].
Despite extensive application of L-hexoses,it is difficult to obtain L-hexoses from natural sources. To meet the demands for L-sugars,it is necessary to develop an efficient method for their synthesis. In recent years scientists have made great efforts in the synthetic approaches toward L-pyranosides. They can be classified into four categories [1],de novo synthesis [9],homologation of shorter monosaccharides [10],elaboration of D-sugars [11] and enzymatic synthesis [12].
Here we report an efficient strategy for the synthesis of L-glucose and L-galactose by using head-to-tail inversion based on the works reported by Li et al. [13]. This strategy takes advantage of the latent symmetrical elements that are present in D-sugars to produce corresponding L-sugars (Scheme 1). Li and co-workers conceived an approach to L-glucose involving the β-C-glycoside prepared from the D-glucosyl acetate using Co2(CO)8-catalyzed silyloxymethylation [13].

|
Download:
|
| Scheme 1.Interchange of groups at C1 and C5. | |
{kind=link}
Several other works with similar strategies have been published: Yang et al. [14],and Doboszewski and Herdewijn [15] had generated L-gulose,L-galactose,L-glucose,and L-allose, using reduction of the aldehyde functional group at C-1 to a primary alcohol and oxidation of the C-6 alcohol to an aldehyde from D-glucose,D-galactose,D-gulose,and D-allose,respectively. Wei et al. [11d, 11f] developed an alternative transformation of 4-deoxypentenosides for the preparation of L-hexoses. And they also achieved the synthesis of the L-galactose derivative through a D-glucal intermediate [11e]. Martı´nez et al. [16] have developed an efficient strategy to convert D-glucose to L-glucose and L-glucuronic acid by periodate cleavage and regiospecific periodate oxidation of the C6-C7.
Differing from Li’s work [13] which could give different orthogonally protected building blocks of L-hexoses containing oligosaccharides from D-glucose,our strategy could give different pentaacetate L-sugars from different D-sugars. The strategy of head-to-tail inversion represents one of the most expedient ways to prepare L-monosaccharides of high optical purity,and should be considered particularly suitable for large scale preparation. Moreover,the strategy can be applied for the assembly of L-hexoses or other rare sugars in biologically relevant oligosaccharide sequences.
The route we developed for the synthesis of L-glucose and L-galactose from D-hexoses utilized the strategy to switch the functional groups at C1 and C5. The strategy was achieved by a one-pot procedure with the oxidation and reduction of the silyl enol ether at C1,and the lead(IV) tetraacetate mediated oxidative decarboxylation at C5. In this case,we achieved a simple and convenient way to synthesize unnatural L-hexoses from the naturally abundant D-hexoses. 2. Experimental 2.1. General
All chemicals were purchased as reagent grade and used without further purification,unless otherwise noted. Dichloromethane was distilled over calcium hydride. THF and toluene were distilled over sodium. Analytical TLC was performed on silica gel 60 F254 precoated on glass plates,with detection by fluorescence and/or by staining with 5% concentrated sulfuric acid in EtOH. Column chromatography was performed on silica gel (230-400 mesh). Optical rotations were measured using a polarimeter at 25 ℃. The 1H NMR spectra were recorded using CDCl3,as solvents at 25 ℃. Chemical shifts (in ppm) were referenced with tetramethylsilane (δ 0) in CDCl3. The 13C NMR spectra were calibrated with CDCl3 (δ 77.16). The positions of hydroxyl groups of mono-ols were determined by the 2D COSY. High-resolution mass spectrum (HRMS) was obtained using electrospray ionization (ESI) mass spectroscopy. Air-sensitive reactions were performed in flame-dried glassware under argon. Organic solvents were evaporated with a rotary evaporator. 2.1.1. General procedure for preparation of ketone 3
Compound 1 (10 mmol) in water (40 mL) was added sodium bicarbonate (15 mmol) and pentane-2,4-dione (12 mmol). After stirring at 90 ℃ for the given time,the reaction was monitored by TLC. After completion,the reaction mixture was washed with CH2Cl2 (50 mL) and treated with Dowex resin (50X8-200,H+ form). The residue was purified by flash chromatography (CHCl3/ MeOH = 10/1) to give product 2. Compound 2 (45.4 mmol) was dissolved in dry pyridine (200 mL) and trityl chloride (54.4 mmol) were added to the solution at 60 ℃. After compound 2 disappeared, acetic anhydride was added to the solution. The solution was stirred overnight at r.t. and then the reaction was quenched by addition of MeOH (60 mL) at 0 ℃. The solvent was removed under reduced pressure and the residue was diluted with CH2Cl2,and successively washed with 1 mol/L HCl(aq), saturated NaHCO3(aq) and brine. The organic layers were dried over Na2SO4. After removal of the solvent under reduced pressure, the residue was purified by flash chromatography (petroleum ether/EtOAc = 4/1,v/v) to give product 3.
3'-(2,3,4-O-Triacetyl-6-O-trityl-β-D-glucopyranosyl)-2'-propanone (3a): [α]D25 +21.5 (c 1.3,CHCl3),yield 85% for 2 steps. 1H NMR (400 MHz,CDCl3): d 7.49-7.12 (m,15H,Ar-H),5.17 (dt,2H,J = 18.2, 9.3 Hz,H-3,H-1),4.95 (t,1H,J = 9.4 Hz,H-4),3.96 (td,1H,J = 9.5, 3.1 Hz,H-2),3.58-3.51 (m,1H,H-5),3.24 (dd,1H,J = 10.5,2.1 Hz, H6a,H6-b),2.80 (dd,1H,J = 15.6,9.2 Hz,-CH2CO-),2.50 (dd,1H, J = 15.6,3.1 Hz,-CH2CO-),2.24 (d,3H,J = 7.8 Hz),2.03 (s,3H),1.98 (s,3H),1.72 (s,3H). 13C NMR (101 MHz,CDCl3): d 205.53,170.38, 169.91,169.01,143.58,128.69,127.77,127.02,86.46,74.45,71.96, 68.74,61.92,45.66,31.32,20.70,20.42. HRMS (ESI) Calcd. for C34H36O9 [M+Na]+: 611.2251; Found: 611.2247.
3'-(2,3,4-O-Triacetyl-6-O-trityl-β-D-mannopyranosyl)-2'-propanone (3b): [α]D25 -8 (c 1,CHCl3),yield 80% for 2 steps. 1H NMR (400 MHz,CDCl3): d 7.18-7.5 (m,15H,Ar-H),5.33 (dd,1H, J = 3.4 Hz,0.7 Hz,H-3),5.32 (t,1H,J = 10.0 Hz,10.4 Hz,H-2),5.04 (dd,1H,J = 10.1,3.4 Hz,H-4),4.18 (dd,1H,J = 8.1,4.6 Hz,H-1),3.54 (ddd,1H,J = 9.9,4.8,2.3 Hz,H-5),3.20 (dd,1H,J = 10.4,2.3 Hz, H-6a),3.07 (dd,1H,J = 10.4,4.9 Hz,H-6b),2.82 (dd,1H,J = 16.4, 8.2 Hz,-CH2CO-),2.47 (d,1H,J = 4.6 Hz,-CH2CO-),2.23 (s,3H), 2.22 (s,3H),1.96 (s,3H),1.73 (s,3H). 13C NMR (101 MHz,CDCl3): d 205.01,170.56,170.15,169.23,143.75,128.73,127.74,126.99, 86.46,77.84,73.01,72.51,70.27,66.24,62.50,44.48,30.84,20.78, 20.66,20.54. HRMS (ESI) Calcd. for C34H36O9 [M+Na]+ 611.2251; Found: 611.2249. 2.1.2. General procedure for preparation of heptitol 7
To a solution of ketone 3 (0.26 mmol) in MeCN-cyclohexane (6:5) (2.2 mL) were added sodium iodide (152 mg),pyridine (73.4 μL),and then TMSCl (116 μL). The solution was stirred under argon overnight at 72 ℃. The reaction was monitored by TLC. After compound 3 disappeared,the solvent was removed under reduced pressure and the residue was diluted with CH2Cl2. Then ozone was bubbled through the solution for 0.5 h at-78 ℃. After compound 4 disappeared,Me2S (0.1 mL) was added and the temperature was raised to room temperature. The reaction was then diluted with CH2Cl2 and successively washed with saturated NaHCO3(aq) and brine. After drying over Na2SO4,NaBH(OAc)3 (276 mg) was added and stirred until compound 5 disappeared. MeOH (0.1 mL) was added to quench the reaction and then washed with saturated NaHCO3(aq) and brine. After drying over Na2SO4,compound 6 was dissolved in dry pyridine. Acetic anhydride was added to the solution and then stirred overnight at r.t. The reaction was quenched by addition of MeOH (1 mL). The residue was purified by flash chromatography (petroleum ether/EtOAc = 6/1,v/v) to give product 7 as semisolids.
1,3,4,5-O-Tetraacetyl-7-O-trityl-2,6-anhydro-D-glycero-Dgluco- heptitol (7a): [α]D25 -8 (c 1,CHCl3),yield 54% for 4 steps. 1H NMR (300 MHz,CDCl3): d 7.43-7.46 (m,6H,Ar-H),7.20-7.32 (m,9H,Ar-H),5.13-5.18 (m,3H,H-3,H-4,H-1),4.27 (d,2H, J = 3.5 Hz,-CH2O-),3.68-3.71 (m,1H,H-2),3.56-3.57 (m,1H,H- 5),3.25 (dd,1H,J = 2.1,10.2 Hz,H-6a),3.07 (dd,1H,J = 4.5,10.5 Hz, H-6b),2.10 (s,3H),2.04 (s,3H),1.99 (s,3H),1.73 (s,3H). 13C NMR (75 MHz,CDCl3): d 170.88,170.64,169.70,169.11,143.74,128.79, 127.85,127.08,86.55,75.65,74.55,68.74,68.63,62.26,62.22, 20.66,20.58,20.55,20.30. HRMS (ESI) Calcd. for C34H36O10 [M+Na]+: 627.2201; Found: 627.2188.
1,3,4,5-O-Tetraacetyl-7-O-trityl-2,6-anhydro-D-glycero-Dmanno- heptitol (7b): [α]D25 +8 (c 1,CHCl3),yield 36% for 4 steps. 1H NMR (400 MHz,CDCl3): d 7.18-7.54 (m,15H,Ar-H),5.44 (d,1H, J = 2.6 Hz,H-3),5.31 (t,1H,J = 10.0 Hz,H-4),5.00 (dd,1H,J = 10.1, 3.3 Hz,H-1),4.27 (dd,1H,J = 11.2,6.7 Hz,-CH2O-),4.12 (m,1H, -CH2O-),3.93 (t,1H,J = 6.6 Hz,H-2),3.56 (ddd,1H,J = 9.9,4.8, 2.4 Hz,H-5),3.16 (qd,2H,J = 10.5,3.7 Hz,H-6),2.19 (s,3H),2.07 (s, 3H),1.97 (s,3H),1.74 (s,3H). 13C NMR (101 MHz,CDCl3): d 170.55, 170.38,170.33,169.16,143.75,128.75,127.77,127.01,86.54, 77.95,74.04,72.43,67.76,66.21,62.65,61.71,20.77,20.70,20.54. HRMS (ESI) Calcd. for C34H36O10 [M+Na]+: 627.2201; Found: 627.2182. 2.1.3. General procedure for preparation of acid 9
The compound 7 (0.8 mmol) was dissolved in dry MeCN,and sodium iodide (2.5 mmol) added,and then TMSCl (2.8 mmol) at 0 ℃. After compound 7 disappeared,the mixture was diluted with CH2Cl2 and washed with brine. The organic layer was dried (Na2SO4),filtered and concentrated to afford compound 8 which was used without further purification.
To a solution of compound 8 (0.12 mmol) in a mixed solvent of water and MeCN (1/10 ratio,3 mL) was consecutively added 2,2,6,6,-tetramethyl-1-piperidinyloxy (0.06 mmol) and iodosobenzene diacetate (0.33 mmol). The reaction was stirred at r.t. and monitored by TLC. After about 4 h,compound 8 disappeared, then the solution was diluted with CH2Cl2,and was sequentially washed with 5% Na2S2O3(aq),saturated KH2PO4(aq). The organic layer was dried over Na2SO4 and the residue was purified by column chromatography (trichloromethane/MeOH = 8/1,v/v) to give product 9.
1,3,4,5-O-Tetraacetyl-2,6-anhydro-D-glycero-D-gluco-heptanoic acid (9a): [α]D25 +40 (c 0.4,CHCl3),yield 85% for 2 steps. 1HNMR (300 MHz,CDCl3): d 8.30 (s,1H,-COOH),5.21-5.32 (m,2H,H-3,H- 1),5.11 (t,1H,J = 9.9 Hz,H-4),4.28 (dd,1H,J = 1.8,12.3 Hz,-CH2O- ),4.18 (dd,1H,J = 12.3 Hz,-CH2O-),4.07 (d,1H,J = 9.2 Hz,H-2), 3.75-3.79 (m,1H,H-5),2.11 (s,3H),2.06 (s,3H),2.04 (s,3H),2.03 (s,3H). 13C NMR (75 MHz,CDCl3): d 171.00,170.42,170.21,169.87, 169.57,75.98,75.51,73.29,68.96,67.80,61.85,20.65,20.48,20.45, 20.41. HRMS (ESI) Calcd. for C15H20O11 [M+Na]+: 399.0898; Found: 399.0888.
1,3,4,5-O-Tetraacetyl-2,6-anhydro-D-glycero-D-manno-heptanoic acid (9b): [α]D25 -8 (c 1,CHCl3),yield 70% for 2 steps. 1H NMR (400 MHz,CDCl3): d 5.47 (d,1H,J = 3.0 Hz,H-3),5.42 (t,1H, J = 10.0 Hz,H-4),5.12 (dd,1H,J = 3.4,10.0 Hz,H-1),4.18 (m,2H, H-2,H-5),4.04 (d,1H,J = 9.9 Hz,H-6a),4.00 (t,1H,J = 6.4 Hz,H-6b), 2.17 (s,3H),2.07 (s,3H),2.06 (s,3H),2.01 (s,3H). 13C NMR (101 MHz,CDCl3): d 170.48,170.17,170.01,169.90,74.68,75.99, 71.40,67.10,66.19,61.54,20.66,20.64,20.62,20.54. HRMS (ESI) Calcd. for C15H20O11 [M+Na]+: 399.0897; Found: 399.0901. 2.1.4. General procedure for preparation of pentaacetate L-sugar 10
Compound 9 (1 mmol) was dissolved under argon atmosphere in a mixture of dry THF (10 mL) and acetic acid (0.1 mL). Lead tetraacetate (6 mmol) was added to the solution and stirred for hours (6 h for 9a and 10 h for 9b) and the solid was removed by filtration. The solution was removed under reduced pressure and the residue was diluted with CH2Cl2,and washed with 1 mol/L HCl(aq),saturated NaHCO3(aq) and brine. The organic layers were dried over Na2SO4 and the solvent was removed under reduced pressure. The residue was purified by column chromatography (petroleum ether/EtOAc = 3:1,v/v) to give product 10.
1,2,3,4,6-O-Pentaacetate-L-glucose (10a): Yield 80%. 1H NMR (400 MHz,CDCl3): d 6.33 (d,2.16 H,J = 3.7 Hz),5.72 (d,1H, J = 8.3 Hz),5.47 (t,2.36 H,J = 9.9 Hz),5.26 (t,1H,J = 9.4 Hz), 5.08-5.17 (m,6.75 H),4.25-4.31 (m,3.41 H),4.08-4.14 (m,5.91 H), 3.82-3.86 (m,1H),2.18,2.12,2.10. 2.09,2.05,2.04,2.03,2.02,2.02 (9s,53H). 13C NMR (100 MHz,CDCl3): d 170.58,170.18,170.05, 169.61,169.35,169.20,168.91,168.70,91.68,89.04,72.76,72.70, 70.21,69.80,69.17,67.87,67.74,61.43,20.83,20.77,20.65,20.62, 20.52,20.40. HRMS (ESI) Calcd. for C16H22O11 [M+Na]+: 413.1054; Found: 413.1061.
1,2,3,4,6-O-Pentaacetate-L-galactose (10b): [α]D25 -16 (c 1, CHCl3),yield 76%. 1H NMR (400 MHz,CDCl3): d 6.38 (d,1H, J = 1.5 Hz,H-1),5.50 (d,1H,J = 1.3 Hz,H-3),5.38-5.29 (m,2H,H-2, H-5),4.35 (td,1H,J = 6.6,0.9 Hz,H-4),4.16-4.03 (m,2H,H-6),2.16 (d,6H,J = 0.6 Hz),2.05 (s,3H),2.02 (s,3H),2.01 (s,3H). 13C NMR (101 MHz,CDCl3): d 170.31,170.09,170.08,169.82,168.86,89.75, 68.78,67.45,67.39,66.47,20.84,20.61,20.60,20.56,20.50. HRMS (ESI) Calcd. for C16H22O11 [M+Na]+: 413.1054; Found: 413.1055. 3. Results and discussion
In our study,we followed the strategy of Li [13] to elongate and shorten the carbon chain at C1 and C5,respectively. There are two key steps in switching the functional groups at C1 and C5. The strategy of head-to-tail inversion has been achieved.
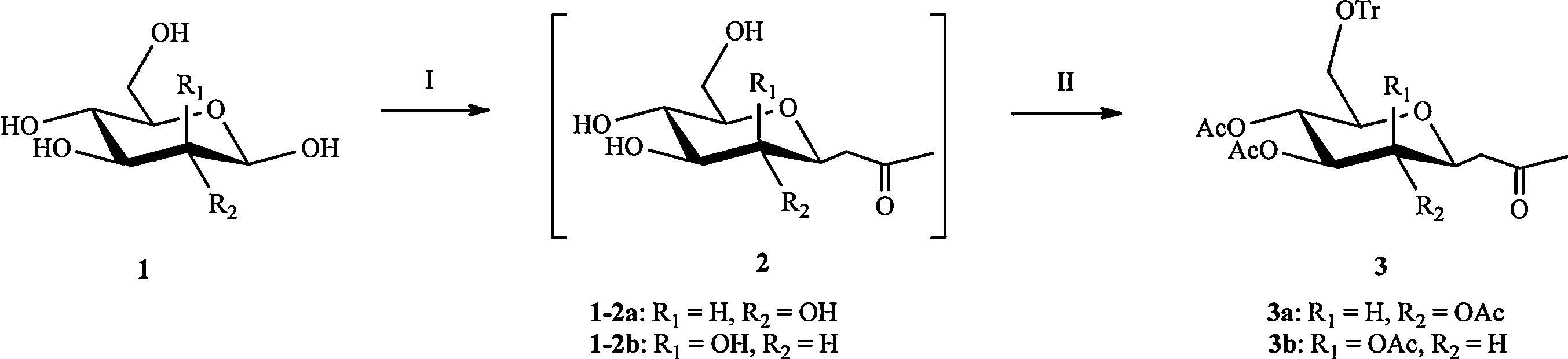
To elongate the carbon chain and introduce a hydroxylmethyl group at C1,a methylene ketone was first synthesized. And then ozone oxidation and followed reduction were used to shorten the carbon chain to a proper length. The details of synthesis are as follows (Scheme 2): The formation of compound 2 was from the initial condensation of the carbanion of the β-diketone with the starting sugar 1,reported by Lubineau et al. [17]. Then,the other hydroxyl groups were protected,including a trityl group introduced to the hydroxyl group at C6 of 2 with trityl chloride,and acetylation of the remaining hydroxyl groups (without purification).

|
Download:
|
| Scheme 2.Synthesis of ketone 3 from D-sugar 1. Reagents and conditions: (Ⅰ) pentane-2,4-dione,NaHCO3,H2O; (Ⅱ) Py,TrCl,Ac2O,85% for 3a,80% for 3b for 2 steps. | |
{kind=link}
Compound 3 was obtained in high overall yields. With ketones 3 in hand,silyl enol ethers were synthesized in order to introduce the hydroxymethyl groups at C-1 by 4 steps (Scheme 3). The reactions were carried out by using the Me3SiCl- NaI-pyridine reagents in MeCN-pentane as described by Norsikian [18],which was performed at 72 ℃ for 12 h with good stereoselectivity. Once obtained,the resulting silyl enol ethers 4 were engaged in an oxidation reaction with ozone at -78 ℃ in CH2Cl2 for 0.5 h. Under these conditions,the aldehydes 5 were obtained, reduced by sodium triacetoxyborohydride,and acetylated to form compounds 7. The elongation at C1 was achieved by this procedure. Since compound 4 was unstable and compound 5 proved difficult to separate,we turned (or proceeded) to a one-pot method to carry out all four steps from compound 3 to compound 7. The one-pot procedure achieved a high yield and the operation was extremely simplified.

|
Download:
|
| Scheme 3.Synthesis of C-glucoside derivative 7. Reagents and conditions: (Ⅰ) TMSCl,NaI,Py,CH3CN-pentane (6/5),(Ⅱ) O3,Me2S,-78℃; (Ⅲ) NaBH(OAc)3; (Ⅳ) Py,Ac2O,54% for 7a,36% for 7b for 4 steps. | |
{kind=link}
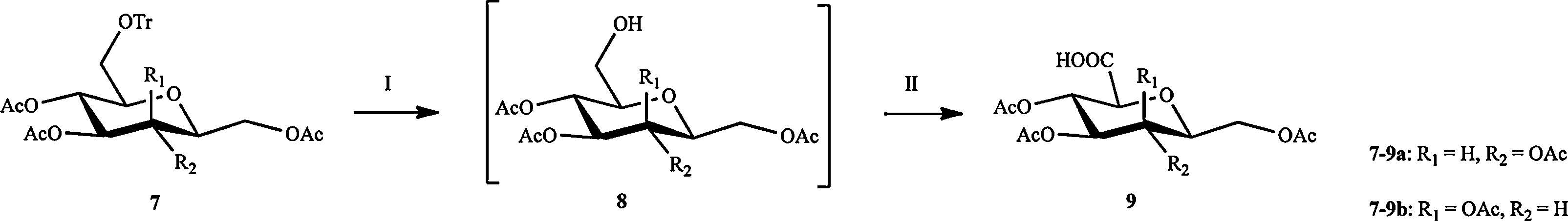
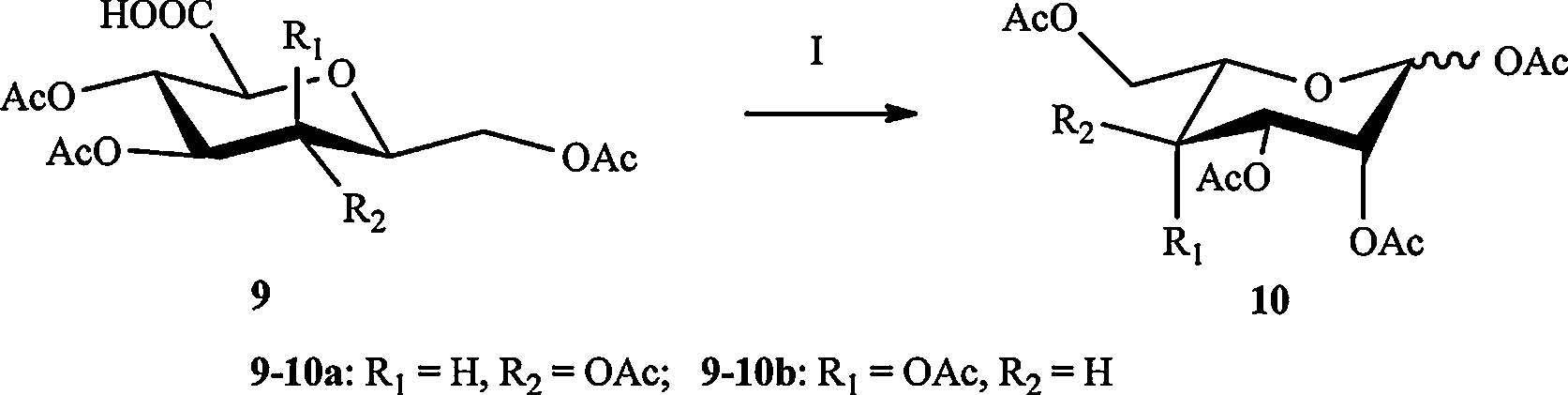
We then switched the methylol group to a hydroxyl group at C5. The subsequent oxidation of primary hydroxyl groups in 8 with 2,2,6,6-tetramethyl-1-piperidinyloxy (TEMPO)/iodosobenzene diacetate (DIB) reagent system [18] provided the carboxylic acid derivatives 9 (Scheme 4). The desired L-sugar derivatives 10 were obtained by oxidative decarboxylation mediated by lead(IV) tetraacetate [19] (Scheme 5). We found that the compound 10a was a mixture of a and b configuration,on the contrary,the compound 10b was in major β configuration. We supposed that this result is due to the different reaction times.

|
Download:
|
| Scheme 4.Synthesis of acid 9. Reagents and conditions: (Ⅰ) TMSCl,NaI,CH3CN; (Ⅱ) TEMPO,DIB,CH3CN-H2O,85% for 9a,70% for 9b for 2 steps. | |
{kind=link}

|
Download:
|
| Scheme 5.Synthesis of L-sugar derivative 10. Reagents and conditions: (Ⅰ) Pb(OAc)4,THF-AcOH (10/1),80% for 10a,76% for 10b. | |
{kind=link}
In summary,we have developed a new and efficient route for the synthesis of L-glucose and L-galactose from L-hexoses with the strategy of head-to-tail inversion in 31% and 15% overall yield. The strategy was achieved by the oxidation and reduction of the silyl enol ether at C1,and lead(Ⅳ) tetraacetate mediated oxidative decarboxylation at C5. It was a convenient and inexpensive way to prepare L-glucose and L-galactose with fewer purification steps, which will facilitate the preparation of oligosaccharides and derivatives containing L-glucose and L-galactose. Acknowledgments
This research was supported by the National Basic Research Program of China (973 Program,No. 2012CB822100),the National Key Technology R&D Program ‘‘New Drug Innovation’’ of China (No. 2012ZX09502001-001) and the National Natural Science Foundation of China (Nos. 20972012 and 21232002).
| [1] | (a) M.M.L. Zulueta, Y.Q. Zhong, S.C. Hung, Synthesis of L-hexoses and their related biomolecules, Chem. Commun. 49 (2013) 3275-3287; (b) D. D'Alonzo, A. Guaragna, G. Palumbo, Recent advances in monosaccharide synthesis: a journey into L-hexose world, Curr. Org. Chem. 13 (2009) 71-98. |
| [2] | Y.S. Li, K. Matsunaga, M. Ishibashi, Y. Ohizumi, Littoralisone, a novel neuritogenic iridolactone having an unprecedented heptacyclic skeleton including four-and nine-membered rings consisting of glucose from Verbena littoralis, J. Org. Chem. 66 (2001) 2165-2167. |
| [3] | (a) Z.J. Segerman, B. Roy, S.M. Hecht, Characterization of bleomycin-mediated cleavage of a hairpin DNA library, Biochemistry 53 (2013) 5315-5327; (b) Z. Yu, R.M. Schmaltz, T.C. Bozeman, R. Paul, M.J. Rishel, Selective tumor cell targeting by the disaccharide moiety of Bleomycin, J. Am. Chem. Soc. 135 (2013) 2883-2886. |
| [4] | T. Ogita, N. Otake, Y. Miyazaki, et al., The structure of adenomycin (C19-97 substance), Tetrahedron Lett. 21 (1980) 3203-3206. |
| [5] | Y. Chen, R.N. Liang, W. Qin, Potentiometric sensor for sensitive and selective detection of heparin, Chin. Chem. Lett. 23 (2012) 233-236. |
| [6] | (a) S. Arungundram, K. Al-Mafraji, J. Asong, et al., Modular synthesis of heparan sulfate oligosaccharides for structure-activity relationship studies, J. Am. Chem. Soc. 131 (2009) 17394-17405; (b) T. Polat, C.H. Wong, Anomeric reactivity-based one-pot synthesis of heparinlike oligosaccharides, J. Am. Chem. Soc. 129 (2007) 12795-12800; (c) U. Lindahl, Heparan sulfate -a polyanion with multiple messages, Pure Appl. Chem. 69 (1997) 1897-1902; (d) D.L. Rabenstein, Heparin and heparan sulfate: structure and function, Nat. Prod. Rep. 19 (2002) 312-331. |
| [7] | (a) P.E. Jansson, B. Lindbery, G. Widmalm, P.A. Sandford, Structural studies of an extracellular polysaccharide (S-130) elaborated by Alcaligenes ATCC 31555, Carbohydr. Res. 139 (1985) 217-223; (b) P.E. Jansson, N.S. Kumar, B. Lindbery, Structural studies of a polysaccharide (S-88) elaborated by Pseudomonas ATCC 31554, Carbohydr. Res. 156 (1986) 165-172; (c) T.A. Chowdhury, B. Lindbery, U. Lindquist, J. Baird, Structural studies of an extracellular polysaccharide (S-198) elaborated by Alcaligenes ATCC 31853, Carbohydr. Res. 161 (1987) 127-132. |
| [8] | (a) W. Kubelka, B. Kop, K. Jentzsch, H. Ruis, Zur biogenese von strophanthidinglykosiden: convallatoxol als vorstufe von convallatoxin in Convallaria majalis, Phytochemistry 13 (1974) 1805-1808; (b) S. Esaki, A. Ohishi, A. Katsumata, N. Sugiyama, S. Kamiya, Synthesis of α-LMannopyranosyl-containing disaccharides and phenols as substrates for the α-L-Mannosidase activity of commercial naringinase, Biosci. Biotechnol. Biochem. 57 (1993) 2099-2103. |
| [9] | (a) M. Takeuchi, T. Taniguchi, K. Ogasawara, Integrated route to the L-aldohexoses using a common man-made chiral building block, Chirality 12 (2000) 338-341; (b) M. Honzumi, T. Taniguchi, K. Ogasawara, A new convergent route to aldohexoses from a common chiral building block, Org. Lett. 3 (2001) 1355-1358; (c) L. Ermolenko, N.A. Sasaki, Diastereoselective synthesis of all eight L-hexoses from L-ascorbic acid, J. Org. Chem. 71 (2006) 693-703; (d) J.M. Harris, M.D. Keranen, G.A. O'Doherty, Syntheses of D-and L-mannose, gulose, and talose via diastereoselective and enantioselective dihydroxylation reactions, J. Org. Chem. 64 (1999) 2982-2983; (e) J.M. Harris, M.D. Keränen, H. Nguyen, V.G. Young, G.A. O'Doherty, Syntheses of four D-and L-hexoses via diastereoselective and enantioselective dihydroxylation reactions, Carbohydr. Res. 328 (2000) 17-36; (f) M.M. Ahmed, G.A. O'Doherty, De novo asymmetric syntheses of D-and L-talose via an iterative dihydroxylation of dienoates, J. Org. Chem. 70 (2005) 10576-10578; (g) W. Du, Y. Hu, Asymmetric synthesis of methyl 6-deoxy-3-O-methyl-α-L-mannopyranoside from a non-carbohydrate precursor, Carbohydr. Res. 341 (2006) 725-729; (h) A.B. Northrup, D.W.C. MacMillan, Two-step synthesis of carbohydrates by selective aldol reactions, Science 305 (2004) 1752-1755; (i) J.Mlynarski, B. Gut, Organocatalytic synthesis of carbohydrates, Chem. Soc. Rev. 41 (2012) 587-596. |
| [10] | (a) A. Guaragna, C. Napolitano, D. D'Alonzo, S. Pedatella, G. Palumbo, A versatile route to L-hexoses: synthesis of L-mannose and L-altrose, Org. Lett. 8 (2006) 4863-4866; (b) A. Guaragna, D. D'Alonzo, C. Paolella, C. Napolitano, G. Palumbo, Highly stereoselective de novo synthesis of L-hexoses, J. Org. Chem. 75 (2010) 3558-3568; (c) L. Ermolenko, N.A. Sasaki, P. Potier, Novel route to L-hexoses from L-ascorbic acid: asymmetric synthesis of L-galactopyranose and L-talopyranose preliminary communication, Helv. Chim. Acta 86 (2003) 3578-3582; (d) A. Dondoni, A. Marra, A. Massi, Carbohydrate homologation by the use of 2-(trimethylsilyl)thiazole. Preparative scale synthesis of rare sugars: L-gulose, L-idose, and the disaccharide subunit of Bleomycin A2, J. Org. Chem. 62 (1997) 6261-6267; (e) T.G. Frihed, M. Heuckendorff, C.M. Pedersen, M. Bols, Easy access to L-mannosides and L-galactosides by using C-H activation of the corresponding 6-deoxysugars, Angew. Chem. Int. Ed. 51 (2012) 12285-12288. |
| [11] | (a) J.C. Lee, S.W. Chang, C.C. Liao, et al., From D-glucose to biologically potent Lhexose derivatives: synthesis of α-L-iduronidase fluorogenic detector and the disaccharide moieties of Bleomycin A2 and heparan sulfate, Chem. Eur. J. 10 (2004) 399-415; (b) H. Takahashi, Y. Hitomi, Y. Iwai, S. Ikegami, A novel and practical synthesis of L-hexoses from D-glycono-1,5-lactones, J. Am. Chem. Soc. 122 (2000) 2995-3000; (c) H. Takahashi, T. Shida, Y. Hitomi, et al., Divergent synthesis of L-sugars and L-iminosugars from D-sugars, Chem. Eur. J. 12 (2006) 5868-5877; (d) F.P. Boulineau, A. Wei, Synthesis of L-sugars from 4-deoxypentenosides, Org. Lett. 4 (2002) 2281-2283; (e) F.P. Boulineau, A. Wei, Conversion of D-glucals into L-glycals and mirror-image carbohydrates, Org. Lett. 6 (2004) 119-121; (f) G. Cheng, R. Fan, J.M. Hernández-Torres, F.P. Boulineau, A.Wei, Syn additions to 4a-epoxypyranosides: synthesis of L-idopyranosides, Org. Lett. 9 (2007) 4849-4852; (g) A.C.Weymouth-Wilson, R.A. Clarkson, N.A. Jones, et al., Large scale synthesis of the acetonides of L-glucuronolactone and of L-glucose: easy access to L-sugar chirons, Tetrahedron Lett. 50 (2009) 6307-6310. |
| [12] | (a) K. Izumori, A strategy for bioproduction of all hexoses, J. Biotechnol. 124 (2006) 717-722; (b) S.M. Dean, W.A. Greenberg, C.H. Wong, Recent advances in aldolase-catalyzed asymmetric synthesis, Adv. Synth. Catal. 349 (2007) 1308-1320. |
| [13] | Y. Li, Z. Yin, B. Wang, X.B. Meng, Z.J. Li, Synthesis of orthogonally protected L-glucose, L-mannose, and L-galactose from D-glucose, Tetrahedron 68 (2012) 6981-6989. |
| [14] | W.B. Yang, S.S. Patil, C.H. Tsai, C.H. Lin, J.M. Fang, The synthesis of L-gulose and L-xylose from D-gluconolactone, Tetrahedron 58 (2002) 253-259. |
| [15] | B. Doboszewski, P. Herdewijn, 1,2,3,4-Di-O-isopropylidene-L-galactose synthesis from its D-enantiomer, Tetrahedron Lett. 53 (2012) 2253-2256. |
| [16] | R.F. Martínez, Z.L. Liu, A.F.G. Glawar, et al., Short and sweet: D-glucose to L-glucose and L-glucuronic acid, Angew. Chem. Int. Ed. 53 (2014) 1160-1162. |
| [17] | F. Rodrigues, Y. Canac, A. Lubineau, A convenient, one-step, synthesis of β-Cglycosidic ketones in aqueous media, Chem. Commun. 20 (2000) 2049-2050. |
| [18] | S. Norsikian, J. Zeitouni, S. Rat, S. Gérard, A. Lubineau, New and general synthesis of β-C-glycosylformaldehydes from easily available β-C-glycosylpropanones, Carbohydr. Res. 342 (2007) 2716-2728. |
| [19] | R.A. Sheldon, J.K. Kochi, Oxidative decarboxylation of acids by lead tetraacetate, Org. React. 19 (1972) 279-421. |