




b Postdoctoral Programme of Chemical Engineering & Technology, Xiangtan University, Xiangtan 411105, China
Diabetes mellitus is a serious worldwide health problem. Each year,the number of patients diagnosed with diabetes mellitus increases. Diabetes mellitus is known to be a lifestyle-related disease,which can cause numerous complications,such as nephropathy,retinopathy,neuropathy,and cardiovascular diseases. Diabetes mellitus is also one of the leading causes of patient mortality. The International Diabetes Federation reported that about 366 million people were diagnosed with diabetes in 2011, and this number is expected to increase to up to 552 million by 2030 [1]. Type II diabetes mellitus is more prevalent in developed countries is characterized by reduced insulin sensitivity and impaired insulin secretion [2, 3, 4, 5, 6]. Type II diabetes mellitus can be effectively treated by α-glucosidase inhibitors,which have the ability to delay and reduce postprandial blood glucose spike [7, 8, 9].
α-Glucosidase is involved in carbohydrate metabolism and has a crucial function in diabetes,viral infection,and cancer. α-glucosidase has diverse bioactivities and is considered an attractive drug target. At present,a number of α-glucosidase inhibitors have been discovered and studied. Anti-diabetic agents that are used in clinical practice,such as acarbose [10],voglibose, and miglitol [11],competitively inhibit α-glucosidase in the brush border of the small intestine,which consequently delay the hydrolysis of carbohydrates and alleviate postprandial hyperglycemia. However,the continuous administration of these agents may cause several adverse effects,such as diarrhea,abdominal discomfort,flatulence [12, 13, 14],and hepatotoxicity [15]. Therefore, developing novel α-glucosidase inhibitors lacking these liabilities is necessary given the therapeutic challenge of type II diabetes mellitus [16, 17, 18, 19, 20, 21, 22]. Flavonoids are widely distributed in plants,for example,vegetables and fruits. Researchers had focused their efforts on a few pharmacological activities of flavonoids,such as, scavenging free radicals and protecting against lipid peroxidation [23, 24]. Recently,researchers showed more interest in some specific flavonoids because of modulating endothelial nitric oxide metabolism and NADPH oxidase activity of them [25, 26, 27, 28, 29, 30, 31]. The results from mechanistic research suggested that flavonoids may also alleviate hyperglycemia,increase insulin secretion,and improve insulin sensitivity [32]. Flavonoids,such as baicalein, luteolin,kaempferol,apigenin,and chrysin,have the ability to inhibit α-glucosidase activity [33, 34]. The structure of the A,B,and C rings are related to the activity of α-glucosidase and α-amylase inhibition. The unsaturated C ring,linkage of the B ring at the 3' position,3-OH,4-CO,and the hydroxyl substitution on the B ring have been reported to increase the inhibitory activity against aglucosidase and α-amylase [35]. In addition,Hakamata et al. [36] have shown that catechin derivatives showed potent antioxidant activity and α-glucosidase inhibitory activity with alkyl side chains of various lengths,whereas Shin et al. [37] have shown that a series of alkyl or acetyl derivatives of chrysin have hypoglycemic effects. Based on the previous studies,we synthesized a series of alkylated flavonoids such as chrysin,diosmetin,apigenin,and luteolin,and studied their α-glucosidase inhibitory activity.
In this study,chrysin,diosmetin,apigenin,and luteolin derivatives were synthesized from the corresponding naturally occurring flavonoids,and their yeast α-glucosidase inhibitory activity were evaluated. We attempted to study the relationship between the structure of the derivatives and their inhibitory effect.
2. Experimental 2.1. Reagents and instrumentsAll solvents used in this research were of analytical grade,and the chemicals used for synthesizing the derivatives were of reagent grade and commercially available. Chrysin,diosmetin,apigenin and luteolin (>98% by HPLC) were purchased from Shanxi Huike Co.,Ltd.,Jiangsu,China,and were used without further purification. Biotech-grade PNPG (4-nitrophenyl-α-D-glucopyranoside),α-glucosidase from baker’s yeast,1-deoxynojirimycin and acarbose were purchased from Sigma Chemical Co.,Ltd. (St. Louis,MO,USA), and L-glutathione (reduced) was obtained from Roche, Switzerland. Nuclear magnetic resonance (NMR) spectra were recorded at 400 MHz for 1H on a Bruker Avance 400 spectrometer in CDCl3 and DMSO-d6 with TMS as an internal standard (chemical shift in ppm,δ). J values are reported in Hertz. Molecular mass was determined by matrix-assisted laser desorption-ionisation timeof- flight mass spectrometry (MALDI-TOF MS) using a Bruker Aupoflex-III mass spectrometer. Elemental analysis (C,H) of the targeted compounds was measured using an elementary Vario EL III analyser. Melting points (mp) of derivatives were determined on a Shanghai SHENGUANG WRS-1B digital melting-point apparatus, China. The absorbance of samples was obtained using a Mapada UV-1600 spectrophotometer,Shanghai,China.
2.2. SynthesisChrysin (1.3 g,5 mmol) in 120 mL of acetone was added to anhydrous potassium carbonate (0.7 g,5 mmol). The mixture was stirred at reflux for 30 min,bromoethane (1.2 mL,15 mmol) was added dropwise,followed by refluxing for 24 h. The mixture was cooled to room temperature,filtered,and was concentrated in vacuo. The residue was purified with a silica gel column eluting with a mixed solvent (EtOAc/CH2Cl2 = 1:10) to obtain O7-ethyl chrysin (5). Yellowish powder; yield: 68.5%; mp: 163.2-163.7 ℃; 1H NMR (400 MHz,CDCl3): δ 12.71 (s,1H,5-OH),7.88 (d,2H, J = 7.9 Hz,aromatic H2',6'),7.53 (d,3H,J = 7.4 Hz,aromatic H3',4',5'), 6.66 (s,1H,aromatic H8),6.49 (s,1H,aromatic H6),6.36 (s,1H, aromatic H3),4.11 (q,2H,J = 7.0 Hz,-OCH2-),1.46 (t,3H,J = 7.0 Hz, -CH3); MALDI-TOF:m/z 283 ([M+H]+); Anal. calcd. for C17H14O4: C, 72.33; H,5.00; found: C,72.10; H,4.99.
Compounds 6 and 7 were obtained according to the method for compound 5 [38].
O7-Butyl chrysin (6): Yellowish powder; yield: 74.5%; mp: 145.9-148.0 ℃; 1H NMR (400 MHz,CDCl3): δ 12.70 (s,1H,5-OH), 7.89 (d,2H,J = 7.7 Hz,aromatic H2',6'),7.53 (d,3H,J = 7.4 Hz, aromatic H3',4',5'),6.66 (s,1H,aromatic H8),6.50 (s,1H,aromatic H3),6.37 (s,1H,aromatic H6),4.05 (t,2H,J = 6.5 Hz,-OCH2-),1.94- 1.68 (m,2H,-CH2-),1.50 (dt,2H,J = 14.7,7.4 Hz,-CH2-),1.00 (t,3H,J = 7.4 Hz,-CH3); MALDI-TOF:m/z 311 ([M+H]+); Anal. calcd. for C19H18O4: C,73.53; H,5.85; found: C,73.42; H,5.82.
O7-Hexyl chrysin (7): Yellowish powder; yield: 89.7%; mp: 143.3-143.6 ℃; 1H NMR (400 MHz,CDCl3): d 12.70 (s,1H,5-OH), 7.89 (d,2H,J = 6.7 Hz,aromatic H2',6'),7.53 (d,3H,J = 7.2 Hz, aromatic H3',4',5'),6.66 (s,1H,aromatic H8),6.50 (s,1H,aromatic H3),6.37 (s,1H,aromatic H6),4.04 (t,2H,J = 6.5 Hz,-OCH2-),1.94- 1.71 (m,2H,-CH2-),1.71-1.32 (m,2H,-CH2-),1.26 (m,4H,- CH2CH2-),0.92 (t,3H,J = 6.4 Hz,-CH3); MALDI-TOF: m/z 339 ([M+H]+); Anal. calcd. for C21H22O4: C,74.54; H,6.55; found: C, 74.24; H,6.41.
General procedures for the synthesis of compounds 8-13 based on the method used to obtain chrysin alkyl derivatives.
O4',O7-Diethyl apigenin (8): Yellowish powder; yield: 62.3%; mp: 159.3-159.4 ℃. 1H NMR (400 MHz,CDCl3): δ 12.80 (s,1H,5- OH),7.82 (d,2H,J = 8.9 Hz,aromatic H2',6'),7.00 (d,2H,J = 8.9 Hz, aromatic H3',5'),6.56 (s,1H,aromatic H8),6.47 (s,1H,aromatic H3), 6.34 (s,1H,aromatic H6),4.11 (dd,4H,J = 6.9,4.1 Hz,-OCH2-),1.45 (t,6H,J = 6.9 Hz,-CH3); MALDI-TOF:m/z 327 ([M+H]+); Anal. calcd. for C19H18O5: C,69.93; H,5.56; found: C,69.73; H,5.49.
O4',O7-Dibutyl apigenin (9): Yellowish powder; yield: 70.6%; mp: 130.9-131.5 ℃. 1H NMR (400 MHz,CDCl3): δ 12.80 (s,1H,5- OH),7.82 (d,2H,J = 8.8 Hz,aromatic H2',6'),7.00 (d,2H,J = 8.8 Hz, aromatic H3',5'),6.56 (s,1H,aromatic H8),6.47 (s,1H,aromatic H3), 6.35 (s,1H,aromatic H6),4.04 (t,4H,J = 6.4 Hz,-OCH2),2.02-1.68 (m,4H,-CH2-),1.68-1.21 (m,4H,-CH2-),0.99 (t,6H,J = 7.3 Hz,- CH3); MALDI-TOF: m/z 383 ([M+H]+); Anal. calcd. for C23H26O5: C, 72.23; H,6.85; found: C,72.14; H,6.83.
O4',O7-Dihexyl apigenin (10): Yellowish powder; yield: 78.4%; mp: 88.5-88.6 ℃; 1H NMR (400 MHz,CDCl3): δ 12.80 (s,1H,5-OH), 7.82 (d,2H,J = 8.8 Hz,aromatic H2',6'),7.00 (d,2H,J = 8.8 Hz, aromatic H3',5'),6.56 (s,1H,aromatic H8),6.47 (s,1H,aromatic H3), 6.35 (s,1H,aromaticH6),4.17-3.93 (m,4H,-OCH2-),2.02-1.66 (m, 4H,-CH2-),1.42 (dd,4H,J = 48.2,3.8 Hz,-CH2-),1.25 (s,8H,- CH2CH2-),0.92 (t,6H,J = 6.7 Hz,-CH3); MALDI-TOF: m/z 439 ([M+H]+); Anal. calcd. for C27H34O5: C,73.94; H,7.81; found: C, 73.79; H,7.79.
O3',O7-Diethyl diosmetin (11): Yellow powder; yield: 55.6%; mp: 191.9-192.2 ℃; 1H NMR (400 MHz,CDCl3): δ 12.78 (s,1H,5- OH),7.51 (d,1H,J = 10.4 Hz,aromatic H60),7.34 (s,1H,aromatic H20),6.98 (d,1H,J = 8.5 Hz,aromatic H5'),6.57 (s,1H,aromatic H8), 6.47 (s,1H,aromatic H3),6.35 (s,1H,aromatic H6),4.15 (dt,4H, J = 30.8,6.9 Hz,-OCH2-),3.96 (s,3H,-OCH3),1.49 (dt,6H,J = 27.5, 7.0 Hz,-CH3); MALDI-TOF: m/z 357 ([M+H]+); Anal. calcd. for C20H20O6: C,67.41; H,5.66; found: C,67.21; H,5.56.
O3',O7-Dibutyl diosmetin (12): Yellow powder; yield: 66.7%; mp: 111.6-112.1 ℃; 1H NMR (400 MHz,CDCl3): δ 12.78 (s,1H,5- OH),7.49 (d,1H,J = 8.4 Hz,aromatic H60),7.34 (s,1H,aromatic H20), 6.96 (d,1H,J = 8.4 Hz,aromatic H5'),6.56 (s,1H,aromatic H8),6.48 (s,1H,aromatic H3),6.35 (s,1H,aromatic H6),4.07 (dt,4H,J = 23.8, 6.3 Hz,-OCH2-),3.94 (s,4H,-OCH3),1.98-1.71 (m,4H,-CH2-), 1.66-1.38 (m,4H,-CH2-),1.12-0.93 (t,6H,-CH3); MALDI-TOF: m/ z 413 ([M+H]+); Anal. calcd. for C24H28O6: C,69.88; H,6.84; found: C,69.58; H,6.62.
O3',O7-Dihexyl diosmetin (13): Yellow powder; yield: 75.9%; mp: 87.4-87.6 ℃; 1H NMR (400 MHz,CDCl3): δ 12.78 (s,1H,5-OH), 7.50 (d,1H,J = 6.7 Hz,aromatic H60),7.34 (s,1H,aromatic H20),6.97 (d,1H,J = 8.5 Hz,aromatic H5'),6.56 (s,1H,aromatic H8),6.48 (s, 1H,aromatic H3),6.35 (s,1H,aromatic H6),4.06 (dt,4H,J = 23.7, 6.6 Hz,-OCH2-),3.94 (s,3H,-OCH3),1.97-1.74 (m,4H,-CH2-), 1.67-1.38 (m,4H,-CH2-),1.36 (m,8H,J = 6.8,3.5 Hz,-CH2CH2-), 0.92 (t,6H,J = 1.4 Hz,-CH3); MALDI-TOF: m/z 469 ([M+H]+); Anal. calcd. for C28H36O6: C,71.77; H,7.74; found: C,71.42; H,7.55.
General procedures for the synthesis of O3',O4'-ethylidene luteolin [30]: Firstly,to a mixture of luteolin (1.5 g,5 mmol) and K2CO3 (0.4 g,2.5 mmol) in 30 mL of DMSO was added 1, 2-dibromoethane dropwise,followed by heating at 70 ℃ for 1 h. The mixture was poured into ice water,filtered,washed with water,the precipitates were dried in vacuo. The residue was chromatographed on silica gel to afford compound 14. Yellow powder; yield: 32.4%; mp: 307.3-308.0 ℃; 1H NMR (400 MHz, DMSO-d6): δ 12.90 (s,1H,5-OH),10.87 (s,1H,7-OH),7.60-7.57 (m, 2H,aromatic H2',6'),7.04-7.02 (d,1H,J = 8 Hz,aromatic H5'),6.87 (s,1H,aromatic H8),6.51 (s,1H,J = 1.7 Hz,aromatic H3),6.20-6.19 (s,1H,J = 4 Hz,aromatic H6),4.33-4.32 (d,4H,J = 4 Hz,- OCH2CH2O-); MALDI-TOF: m/z 313 ([M+H]+).
General procedures for synthesis of compounds 15-17 based on the method used to obtain chrysin alkyl derivatives.
O7-Ethyl-O3',O4'-ethylidene luteolin (15): Yellow powder; yield: 70.2%; mp: 174.1-174.6 ℃; 1H NMR (400 MHz,CDCl3): δ 7.41-7.37 (m,2H,aromatic H2',6'),6.98-6.96 (d,1H,J = 8 Hz, aromatic H5'),6.54 (s,1H,aromatic H8),6.45 (s,1H,aromatic H3), 6.34 (s,1H,aromatic H6),4.33-4.32 (d,4H,J = 4 Hz,-CH2CH2-), 4.13-4.08 (q,2H,J = 6.7 Hz,-CH2-),1.47-1.43 (t,3H,J = 8 Hz,- CH3); MALDI-TOF: m/z 341 ([M+H]+); Anal. calcd.Anal. calcd. for C19H10O6: C,67.05; H,4.74; found: C,67.25; H,4.79.
O7-Butyl-O3',O4'-ethylidene luteolin (16): Yellow powder; yield: 80.4%; mp: 153.9-154.5 ℃; 1H NMR (400 MHz,CDCl3): δ 7.41-7.37 (m,2H,aromatic H2',6'),6.98-6.96 (d,1H,J = 8 Hz, aromatic H5'),6.54 (s,1H,aromatic H8),6.45 (s,1H,aromatic H3), 6.34 (s,1H,aromatic H6),4.33-4.32 (d,4H,J = 4 Hz,-CH2CH2-), 4.04-4.01 (t,2H,J = 6.2 Hz,-CH2O-),1.81-1.78 (t,2H,J = 7 Hz,- CH2-),1.53-1.47 (q,2H,J = 7.2 Hz,-CH2-),1.01-0.97 (t,3H, J = 7.2 Hz,-CH3); MALDI-TOF: m/z 369 ([M+H]+); Anal. calcd. for C21H20O6: C,68.47; H,5.47; found: C,68.35; H,5.50.
O7-Hexyl-O3',O4'-ethylidene luteolin (17): Yellow powder; yield: 75.9%; mp: 125.6-125.9 ℃. 1H NMR (400 MHz,CDCl3): δ 7.40-7.36 (m,2H,aromatic H2',6'),6.97-6.95 (d,1H,J = 8 Hz, aromatic H5'),6.52 (s,1H,aromatic H8),6.44 (s,1H,aromatic H3), 6.33 (s,1H,aromatic H6),4.32-4.31 (d,4H,J = 4 Hz,-CH2CH2-), 4.03-4.00 (t,2H,J = 6.5 Hz,-OCH2-),1.82-1.78 (t,2H,J = 8 Hz,- CH2-),1.46-1.34 (m,6H,-CH2CH2CH2-),0.91 (s,3H,-CH3); MALDI-TOF: m/z 397 ([M+H]+); Anal. calcd. for C23H24O6: C,69.68; H,6.10; found: C,69.45; H,6.15.
2.3. α-Glucosidase inhibitory activityAn improved methodology used in the assessment of α-glucosidase inhibitory activity has been reported in previous studies [39, 40, 41, 42]. pNP-α-Glu was hydrolyzed into pNP by α-glucosidase. The absorption of pNP at 400 nm was determined using a spectrophotometer,which indicated the activity of α-glucosidase. Sodium phosphate buffer (pH 6.8),50 μL of the samples in various concentrations,and α-glucosidase (200 μL, 0.3 U/mL) were mixed at 37 ℃ and the mixture was allowed to stir for 10 min. The reaction was started with the addition of 4- nitrophenyl-α-D-glucopyranoside (500 μL,10 μmol/L) at 37 ℃ and reaction lasted for 20 min. An aliquot (200 μL) was added to a sodium carbonate solution (9.8 mL,100 μmol/L) to stop the reaction. The glucosidase activity was detected using a silica dish, and the absorbance was determined at 400 nm (for pNP). The inhibitory degree (%) of the targeted compounds upon α-glucosidase addition was calculated using [(1 ΔAtest/ ΔAcontrol) × 100],where ΔA indicates the absorbance increase in 20 min. The 50% inhibitory concentration values (IC50) were expressed as mean ± SD (n = 3),which were obtained using the Sigmaplot 12.0 software. All the tests were performed in triplicate. Acarbose and 1-deoxynojirimycin were used as positive controls.
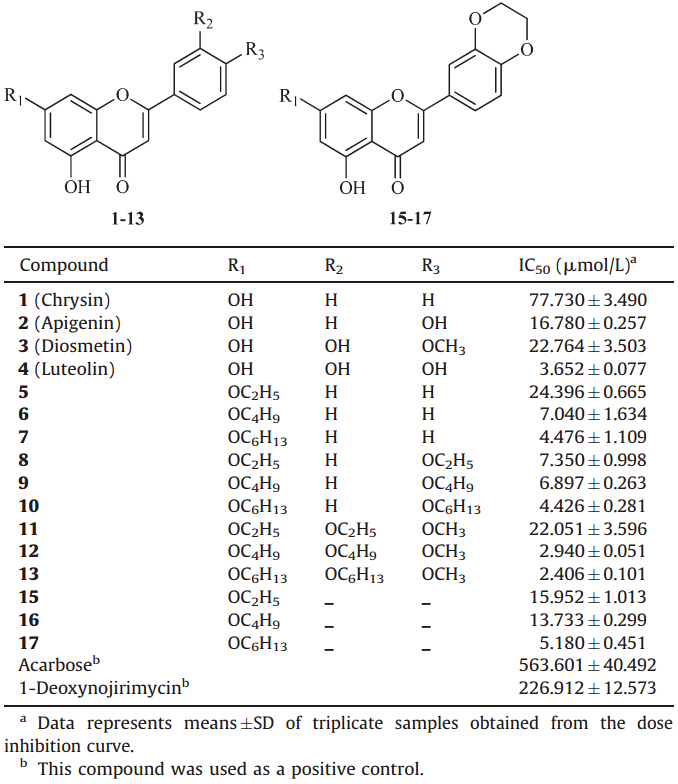
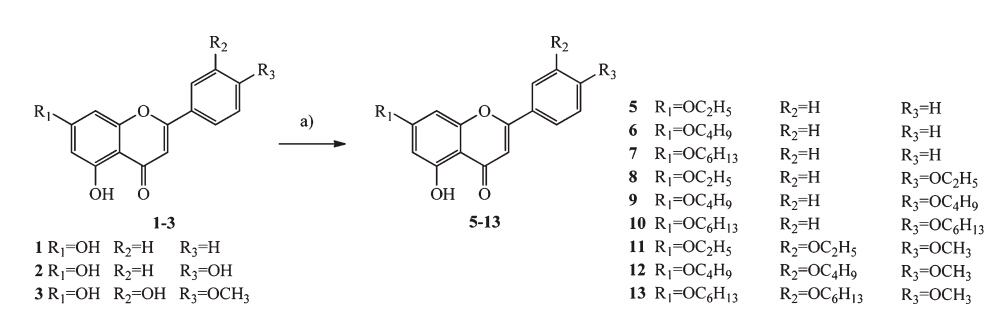
3. Results and discussionThe synthesis of derivatives 5-13 was achieved by coupling various bromoalkanes with their respective precursors in anhydrous acetone (Scheme 1). Chrysin derivatives 5-7 were derived at the C-7 position,diosmetin derivatives 8-10 were derived at the C-7 and C-3' positions,and apigenin derivatives 11-13 were obtained by alkylating the C-7 and C-4' positions. Luteolin derivatives 15-17 were conveniently prepared using the method described in literature (Scheme 2). The structures of all the derivatives were characterized by 1H NMR,MALDI-TOF,and elemental analyses.

|
Download:
|
| Scheme 1.Preparation of derivatives 5-13,reagents and conditions: (a) bromoalkane,K2CO3,acetone,reflux. | |

|
Download:
|
| Scheme 2.Preparation of derivatives 15-16,reagents and conditions: (a) dibromoethane,K2CO3,DMSO,70 ℃; (b) bromoalkane,K2CO3,acetone,reflux. | |
The α-glucosidase inhibition effect of the precursors and their derivatives were determined using similar protocols described in literature [39, 43, 44]. 1-Deoxynojirimycin (a clinically used aglucosidase inhibitor) and acarbose were used as reference drugs. The IC50 values of all compounds are shown in Table 1,which shows that the precursors 1-4 and their derivatives 5-13 and 15-17 were more active against yeast α-glucosidase than 1- deoxynojirimycin (IC50 = 226.912 ± 12.573 μmol/L) and acarbose (IC50 = 563.601 ± 40.492 μmol/L). The α-glucosidase inhibition of chrysin 1 (IC50 = 77.730 ± 3.490 μmol/L) was weaker compared with its derivatives 5-7 (IC50 < 24.396 μmol/L). Similarly,apigenin derivatives 8-10 (IC50 < 7.350 μmol/L) and diosmetin derivatives 11-13 (IC50 < 22.051 μmol/L) are more potent that the parent apigenin 2 (IC50 = 16.780 ± 0.257 μmol/L) and diosmetin 3 (IC50 = 22.764 ± 3.503 μmol/L). The inhibitory activity of luteolin derivatives 15-17 did not exceed that of luteolin 4. But increasing the hydrophobicity or the alkyl chain length led to more potent analogs (IC50: 15 > 16 > 17).
| Table 1 Structures and in vitro α-glucosidase inhibitory activity of targeted compounds. |
{kind=link}
{kind=link}
Transition states of the reactions catalyzed by glycosidase involve covalent intermediates [45, 46]. Hydrogen bonding is a crucial factor for the interactions between the enzyme and its substrates and the conformation and orientations of the inhibitors in the active site [47]. Previous studies have reported that the hydroxyl group has an important function in α-glucosidase inhibition at position 5 of the flavonoids [48, 49],and the hydroxyl substitution on the B ring by particular groups could enhance inhibitory activity [35]. Compared with that of chrysin 1 (IC50 = 26.417 ± 0.485 μmol/L),the inhibitory effect of the 7- methylated compound 5 (IC50 = 24.396 ± 0.665 μmol/L) was minimally increased,whereas butylated and hexylated analogs 6 and 7 (6: IC50 = 7.040 ± 1.634 μmol/L; 7: IC50 = 4.476 ± 1.109 μmol/L) showed excellent activity against the yeast α-glucosidase (Fig. 1). Therefore,α-glucosidase inhibitory activitywas improvedwhen the hydrophobicity of chrysin was enhanced. To further study the effect of flavone hydrophobicity on inhibitory activity,we analyzed the inhibitory activity of apigenin derivatives (IC50: 8 > 9 > 10) and diosmetin derivatives (IC50: 11 > 12 > 13) from Fig. 1. The results indicated that hydrophobicity has an important function in increasing inhibitory activity. Inhibitory activity was dramatically enhanced when the alkyl chain was longer than or equal to two carbons. In addition,We found that the OH groups at positions 30 and 40 of benzopyran that were alkylated by ethylidene did not improve α-glucosidase inhibitory activity; the inhibitory activity of luteolin derivatives 15-17 did not exceed that of luteolin 4. But increasing the hydrophobicity or the elongation of alkyl chains improved the inhibitory activity (IC50: 15 > 16 > 17).

|
Download:
|
| Fig. 1. Effect of compounds on α-glucosidase activity in vitro. | |
{kind=link}
Kumar et al. [49] reported that replacement of the OH group at positions 3'.4' and 7 of benzopyran by any electron-withdrawing group (NO2,OCH3 etc.) favors α-glucosidase inhibitory activity. Therefore,the substitution of the hydrogen of OH by alkyls at position 7 of benzopyran would enhance the inhibitory activity, which is dependent on the alkyl chain lengths. The lengths were found to be positively correlated with the IC50 values of derivatives (IC50: 5 > 6 > 7,8 > 9 > 10,11 > 12 > 13,15 > 16 > 17). To develop novel α-glucosidase inhibitor,the results of this study could provide a series of potential compounds.
4. ConclusionIn summary,twelve chrysin,diosmetin,apigenin,and luteolin alkyl derivatives were synthesized as novel inhibitors of yeast aglucosidase. All compounds were verified by 1H NMR,MALDI-TOF, and elemental analyses. The glucosidase inhibitory activity of all derivatives is higher compared with those of the positive control drugs,acarbose,and 1-deoxynojirimycin. O7-Hexyl chrysin 7, O4',O7-hexyl apigenin 10,O3',O7-hexyl diosmetin 13,O7-hexyl- O3',O4'-ethylidene luteolin 17 were the most effective among the inhomogeneous derivatives. From a structural-activity relationship perspective,replacing the hydrogen of OH group at positions 3',4',and 7 of benzopyran with various alkyl chains was the critical factor in altering the inhibition activity of flavonoids. To develop novel α-glucosidase inhibitor,the results of this study could provide a series of potential compounds.
AcknowledgmentsThis work was financially supported by the Research Fund for the Doctoral Program of Higher Education of China (No. 20114301120004),Hunan Provincial Natural Science Foundation of China (No. 12JJ6081),Dr.’s Start-up Foundation of Xiangtan University (No. 06KZjKZ08035),and Natural Science Foundation of Xiangtan University (No. 2011XZX09).
| [1] | D.R. Whiting, L. Guariguata, C. Weil, J. Shaw, IDF diabetes atlas: global estimates of the prevalence of diabetes for 2011 and 2030, Diabetes Res. Clin. Pract. 94 (2011) 311-321. |
| [2] | S.I. Taylor, D. Accili, Y. Imai, Insulin resistance or insulin deficiency: which is the primary cause of NIDDM? Diabetes 43 (1994) 735-740. |
| [3] | D. Porte Jr., β-Cells in type II diabetes mellitus, Diabetes 40 (1991) 166-180. |
| [4] | A.E. Butler, J. Janson, S. Bonner-Weir, et al., β-Cell deficit and increased β-cell apoptosis in humans with type 2 diabetes, Diabetes 52 (2003) 102-110. |
| [5] | P.C. Tang, Z.G. Lin, Y. Wang, et al., Design and synthesis of DPP-4 inhibitor for the treatment of type 2 diabetes, Chin. Chem. Lett. 21 (2010) 253-256. |
| [6] | Y.H. Wu, Synthesis of (S)-2-ethoxy-3-phenylpropanoic acid derivatives and their insulin-sensitizing activity, Chin. J. Chem. 25 (2007) 265-267. |
| [7] | A.H. Samad, T.S.T. Willing, K.G.M. Alberti, R. Taylor, Effects of BAYm 1099, new aglucosidase inhibitor, on acute metabolic responses and metabolic control in NIDDM over 1 mo, Diabetes Care 11 (1988) 337-344. |
| [8] | N. Asano, Glycosidase inhibitors: update and perspectives on practical use, Glycobiology 13 (2003) 93R-104R. |
| [9] | K. O'Dea, J. Turton, Optimum effectiveness of intestinal alpha-glucosidase inhibitors: importance of uniform distribution through a meal, Am. J. Clin. Nutr. 41 (1985) 511-516. |
| [10] | P. Lefebvre, A. Scheen, The use of acarbose in the prevention and treatment of hypoglycaemia, Eur. J. Clin. Invest. 24 (1994) 40-44. |
| [11] | L.J. Scott, C.M. Spencer, Miglitol: a review of its therapeutic potential in type 2 diabetes mellitus, Drugs 59 (2000) 521-549. |
| [12] | L.K. Campbell, D.E. Baker, R.K. Campbell, Miglitol: assessment of its role in the treatment of patients with diabetes mellitus, Ann. Pharmacother. 34 (2000) 1291-1301. |
| [13] | A.J. Krentz, C.J. Bailey, Oral antidiabetic agents, Drugs 65 (2005) 385-411. |
| [14] | D. Nathan, J. Buse, M. Davidson, et al., Management of hyperglycaemia in type 2 diabetes: a consensus algorithm for the initiation and adjustment of therapy, Diabetologia 49 (2006) 1711-1721. |
| [15] | S.H. Hsiao, L.H. Liao, P.N. Cheng, T.J. Wu, Hepatotoxicity associated with acarbose therapy, Ann. Pharmacother. 40 (2006) 151-154. |
| [16] | Z.Y. Du, R.R. Liu, W.Y. Shao, et al., α-Glucosidase inhibition of natural curcuminoids and curcumin analogs, Eur. J. Med. Chem. 41 (2006) 213-218. |
| [17] | E.B. de Melo, A. da Silveira Gomes, I. Carvalho, α-and β-glucosidase inhibitors: chemical structure and biological activity, Tetrahedron 62 (2006) 10277-10302. |
| [18] | Y.I. Kwon, E. Apostolidis, K. Shetty, In vitro studies of eggplant (Solanum melongena) phenolics as inhibitors of key enzymes relevant for type 2 diabetes and hypertension, Bioresour. Technol. 99 (2008) 2981-2988. |
| [19] | R. Tundis, M. Loizzo, F. Menichini, Natural products as-amylase and-glucosidase inhibitors and their hypoglycaemic potential in the treatment of diabetes: an update, Mini-Rev. Med. Chem. 10 (2010) 315-331. |
| [20] | L.G. Ranilla, Y.I. Kwon, E. Apostolidis, K. Shetty, Phenolic compounds, antioxidant activity and in vitro inhibitory potential against key enzymes relevant for hyperglycemia and hypertension of commonly used medicinal plants, herbs and spices in Latin America, Bioresour. Technol. 101 (2010) 4676-4689. |
| [21] | M. Liu, W. Zhang, J. Wei, X. Lin, Synthesis and α-glucosidase inhibitory mechanisms of bis(2, 3-dibromo-4, 5-dihydroxybenzyl) ether, a potential marine bromophenol α-glucosidase inhibitor, Mar. Drugs 9 (2011) 1554-1565. |
| [22] | R.R. Rao, A.K. Tiwari, P.P. Reddy, et al., Synthesis of antihyperglycemic, α-glucosidase inhibitory, and DPPH free radical scavenging furanochalcones, Med. Chem. Res. 21 (2012) 760-774. |
| [23] | J.D. Xu, L.W. Zhang, Y.F. Liu, Synthesis and antioxidant activities of flavonoids derivatives, troxerutin and 3', 4', 7-triacetoxyethoxyquercetin, Chin. Chem. Lett. 24 (2013) 223-226. |
| [24] | J.B. Zheng, H.F. Zhang, H. Gao, Investigation on electrochemical behavior and scavenging superoxide anion ability of chrysin at mercury electrode, Chin. J. Chem. 23 (2005) 1042-1046. |
| [25] | H.D. Ly, S.G. Withers, Mutagenesis of glycosidases, Annu. Rev. Biochem. 68 (1999) 487-522. |
| [26] | T. Schewe, Y. Steffen, H. Sies, How do dietary flavanols improve vascular function? A position paper, Arch. Biochem. Biophys. 476 (2008) 102-106. |
| [27] | M.N. Clifford, Chlorogenic acids and other cinnamates -nature, occurrence and dietary burden, J. Sci. Food Agric. 79 (1999) 362-372. |
| [28] | M. Richelle, I. Tavazzi, E. Offord, Comparison of the antioxidant activity of commonly consumed polyphenolic beverages (coffee, cocoa, and tea) prepared per cup serving, J. Agric. Food Chem. 49 (2001) 3438-3442. |
| [29] | A. Crozier, I.B. Jaganath, M.N. Clifford, Dietary phenolics: chemistry, bioavailability and effects on health, Nat. Prod. Rep. 26 (2009) 1001-1043. |
| [30] | Q.Q. Wang, N. Cheng, X.W. Zheng, S.M. Peng, X.Q. Zou, Synthesis of organic nitrates of luteolin as a novel class of potent aldose reductase inhibitors, Bioorg. Med. Chem. 21 (2013) 4301-4310. |
| [31] | J.H. Cui, D. Hu, X. Zhang, Z. Jing, et al., Design and synthesis of new 7, 8-dimethoxya-naphthoflavones as CYP1A1 inhibitors, Chin. Chem. Lett. 24 (2013) 215-218. |
| [32] | K. Hanhineva, R. Törrönen, I. Bondia-Pons, et al., Impact of dietary polyphenols on carbohydrate metabolism, Int. J. Mol. Sci. 11 (2010) 1365-1402. |
| [33] | T. Nishioka, J. Kawabata, Y. Aoyama, Baicalein, an a-glucosidase inhibitor from Scutellaria baicalensis, J. Nat. Prod. 61 (1998) 1413-1415. |
| [34] | H.W. Ryu, B.W. Lee, M.J. Curtis-Long, et al., Polyphenols from Broussonetia papyrifera displaying potent a-glucosidase inhibition, J. Agric. Food Chem. 58 (2009) 202-208. |
| [35] | K. Tadera, Y. Minami, K. Takamatsu, T. Matsuoka, Inhibition of α-glucosidase and a-amylase by flavonoids, J. Nutr. Sci. Vitaminol. (Tokyo) 52 (2006) 149-153. |
| [36] | W. Hakamata, I. Nakanishi, Y. Masuda, et al., Planar catechin analogues with alkyl side chains: a potent antioxidant and an α-glucosidase inhibitor, J. Am. Chem. Soc. 128 (2006) 6524-6525. |
| [37] | J.S. Shin, K.S. Kim, M.B. Kim, J.H. Jeong, B.K. Kim, Synthesis and hypoglycemic effect of chrysin derivatives, Bioorg. Med. Chem. Lett. 9 (1999) 869-874. |
| [38] | D.C. Wan, J.J. Yuan, Z.L. Yang, et al., Facile O-alkylation of highly hydrophilic hyperbranched polyglycerol, Chin. Chem. Lett. 18 (2007) 192-194. |
| [39] | Y. Kashima, H. Yamaki, T. Suzuki, M. Miyazawa, Structure-activity relationships of bergenin derivatives effect on α-glucosidase inhibition, J. Enzyme Inhib. Med. Chem. 28 (2013) 1162-1170. |
| [40] | Y.Q. Li, F.C. Zhou, F. Gao, J.S. Bian, F. Shan, Comparative evaluation of quercetin, isoquercetin and rutin as inhibitors of α-glucosidase, J. Agric. Food. Chem. 57 (2009) 11463-11468. |
| [41] | W. Li, K. Wei, H. Fu, K. Koike, Structure and absolute configuration of clerodane diterpene glycosides and a rearranged cadinane sesquiterpene glycoside from the stems of Tinospora sinensis, J. Nat. Prod. 70 (2007) 1971-1976. |
| [42] | S. Adisakwattana, P. Charoenlertkul, S. Yibchok-anun, α-Glucosidase inhibitory activity of cyanidin-3-galactoside and synergistic effect with acarbose, J. Enzyme Inhib. Med. Chem. 24 (2009) 65-69. |
| [43] | C.M. Ma, M. Hattori, M. Daneshtalab, L. Wang, Chlorogenic acid derivatives with alkyl chains of different lengths and orientations: potent α-glucosidase inhibitors, J. Med. Chem. 51 (2008) 6188-6194. |
| [44] | G.L. Li, J.Y. He, A. Zhang, et al., Toward potent α-glucosidase inhibitors based on xanthones: a closer look into the structure-activity correlations, Eur. J. Med. Chem. 46 (2011) 4050-4055. |
| [45] | T.D. Heightman, A.T. Vasella, Recent insights into inhibition, structure, and mechanism of configuration-retaining glycosidases, Angew. Chem. Int. Ed. 38 (1999) 750-770. |
| [46] | A. Vasella, G.J. Davies, M. Böhm, Glycosidase mechanisms, Curr. Opin. Chem. Biol. 6 (2002) 619-629. |
| [47] | D.L. Zechel, S.G. Withers, Glycosidase mechanisms: anatomy of a finely tuned catalyst, Acc. Chem. Res. 33 (2000) 11-18. |
| [48] | H. Gao, T. Nishioka, J. Kawabata, T. Kasai, Structure-activity relationships for aglucosidase inhibition of baicalein, 5, 6, 7-trihydroxyflavone: the effect of A-ring substitution, Biosci. Biotechnol. Biochem. 68 (2004) 369-375. |
| [49] | V. Kumar, S. Kumar, P. Rani, Pharmacophore modeling and 3D-QSAR studies on flavonoids as a-glucosidase inhibitors, Der Pharma Chemica 2 (2010) 324-335. |