




b Beijing Key Laboratory of Active Substance Discovery and Druggability Evaluation, Institute of Materia Medica, Peking Union Medical College and Chinese Academy of Medical Sciences, Beijing 100050, China
Justicidin G is a natural aryl-naphthalene lignan lactone isolated from the plant of Justicia procumbens L.,which has shown significant antitumor activity [1, 2, 3]. Justicidin G has a similar chemical structure to that of etoposide and teniposide,but displays stronger anticancer activity and fewer side effects and is now being further investigated as a new drug development candidate [4, 5, 6, 7]. Considering the poor solubility of justicidin G and the successful structural modification experience of etoposide phosphate ester [8],we made an attempt to synthesis the glycosylated and phosphate ester derivatives of justicidin G. However,the product yields of both derivatives were very low. Moreover,our previous study of justicidinoside B (the glycosylated product of justicidin G) indicated that derivatives with substituents at the 60-OH position exhibited hindered rotation around the aryl-naphthalene bond,which led to difficult structural modifications and poor chemical stability [9, 10]. Hence,we have further investigated the novel justicidin G analogue 13 with a 50-OH group and its phosphate ester 15.Compounds 10-15 were evaluated for their in vitro cytotoxicity against a panel of human tumour cell lines. 2. Experimental
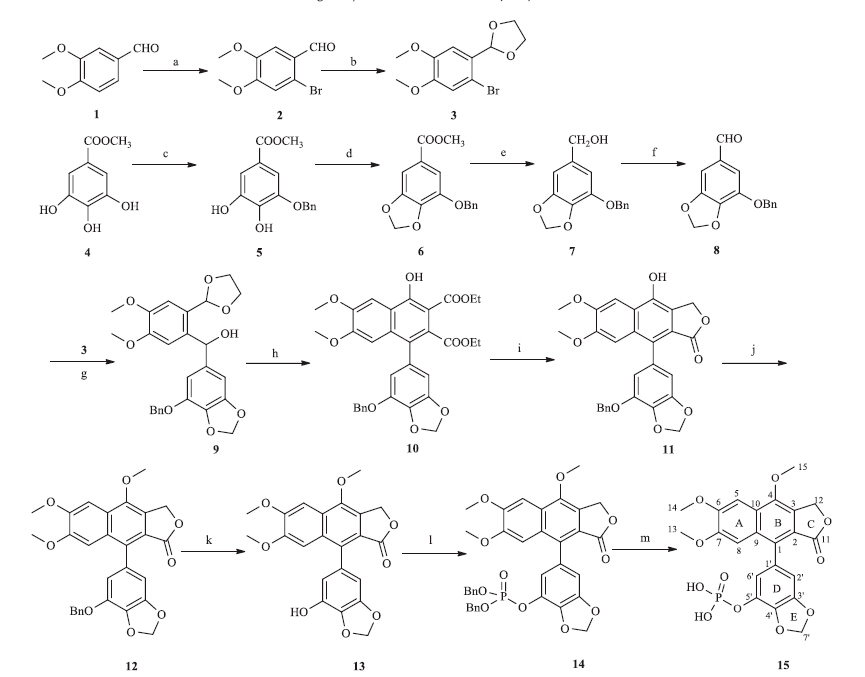
The synthesis of novel justicidin G analogue 13 and its phosphate ester 15 were realized in several steps starting from commercially available methyl gallate and veratraldehyde (Scheme 1). Compound 3 was synthesized in two steps from veratraldehyde in 90% yield [11]. Compound 8 was obtained in four steps from methyl gallate in 81% yield [12]. Treatment of compound 3 with BuLi and Compound 8 under nitrogen atmosphere at -78°C afforded Compound 9 in 70% yield [9]. The synthesis of Compound 10 was accomplished via a Diels-Alder reaction from Compound 9 and DEADC under acidic conditions [3]. Compound 10 was reduced with NaBH4 in tetrahydrofuran under refluxing to afford Compound 11 in 92% yield [10]. Methylation of Compound 11 with CH3I/K2CO3 in acetone under refluxing yielded Compound 12 [9]. Compound 13 was prepared by treatment of the Compound 12 with 10% Pd/C in methanol at ambient temperature. Treatment of Compound 13 with EtN(i-Pr)2/CCl4/4-DMAP/ HPO(OBn)2 in acetonitrile at -30°C afforded compound 14 in 92% yield [8]. Compound 15 was synthesized by treatment of the compound 14 with 10% Pd/C in methanol at ambient temperature.All these compounds were characterized by NMR and HR-ESI-MS analyses [13, 14, 15].

|
Download:
|
| Scheme 1.Synthesis of justicidin G analogue and its phosphate ester. Conditions and reagents: (a) Br2,CH3OH,r.t.,12 h,93%; (b) ethanediol,TsOH·H2O,toluene,reflux,3 h, 96%; (c) NaH,B(OCH3)3,BnBr,DMF,6 h,92%; (d) CH2Br2,DMF,110°C,1 h,96%; (e) LiAlH4,THF,60°C,4 h,93%; (f) PCC,CH2CI2,3 h,95%; (g) n-BuLi,THF,-78°C,6 h,70%; (h) DEADC,AcOH,CH2CI2,140°C,12 h,46%; (i) NaBH4,THF,reflux,3 h,92%; (j) CH3I,K2CO3,acetone,reflux,1 h,96%; (k) H2,10% Pd/C,CH3OH,r.t.,3 h,97%; (l) EtN(i-Pr)2,4-DMAP, CCl4,HPO(OBn)2,CH3CN,-30°C,5 h,92%; (m) H2,10% Pd/C,CH3OH,r.t.,3 h,93%. | |
n Herein,we report a novel and facile synthesis of compounds 13 and 15 in good yields. Compound 10 was a key intermediate for the total synthesis of Compound 15. In a previous study,the reproducibility of the step converting 9 to 10 was less than desirable. In this work we optimized the conditions for this reaction and screened different solvents,amounts of acidic reagent,reaction temperatures and time. We found that the amount of the acidic reagent and reaction temperature were the main parameters affecting the yield of the production of compound 10. The optimal reaction conditions were found to be 2-3 equiv. of acetic acid under 140°C in an appropriate volume of methylene chloride,whereas excessive solvent will reduce the product yield.
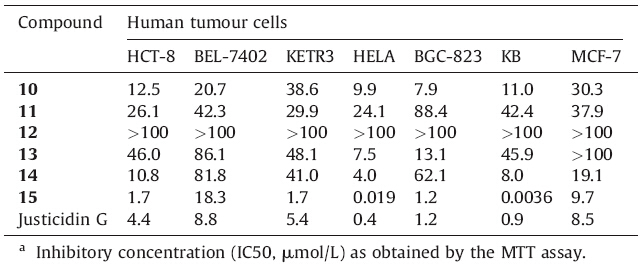
The cytotoxicity of compounds 10-15 was tested against HCT-8,BEL-7402,KETR3,HELA,BGC-823,KB and MCF-7 cell lines by the MTT method. The results are summarized in Table 1. Justicidin G was used as a control compound. From the screening results,it is evident that all compounds except Compound 12 exhibited some level of activity against these cell lines. HCT-8, HELA and KB were more sensitive than BEL-7402 and KETR3 to the justicidin G analogues. It is noteworthy that Compound 15 as a phosphate ester not only possessed better water-solubility (3.5 g/100 mL) [16],but also raised its cytotoxicity against HELA and KB to nanomolar level.
| Table 1 Cytotoxicity of compounds 10-15 against human tumour cells (IC50,μmol/L).a |
{kind=link}
In conclusion,we have synthesized a novel justicidin G analogue 13 with a 50-OH group and its phosphate ester 15 for the first time,and their structures were characterized by NMR spectroscopy and HR-ESI-MS. The cytotoxicity of compounds 10-15 was evaluated in seven cell lines by the MTT method and the results showed that five of them exhibited good antitumor activity against these cell lines. Moreover,the cytotoxicity of Compound 15 against HELA and KB was improved to nanomolar level. This result is helpful in discovering new antitumor agents. Acknowledgments
We thank National Major Scientific and Technological Special Project for ‘Significant New Drugs Innovation’ (No. 2012ZX09301002- 001),the Union Youth Science & Research Fund (No. 3332013074) for financial support.
| [1] | C.C. Chen, W.C. Hsin, Y.L. Huang, Six new diarylbutane lignans from Justicia procumbens, J. Nat. Prod. 61 (1998) 227-229. |
| [2] | M.H. Yang, J. Wu, F. Cheng, Y. Zhou, Complete assignments of 1H and 13C NMR data for seven arylnaphthalide lignans from Justicia procumbens, Magn. Reson. Chem. 44 (2006) 727-730. |
| [3] | X.L. He, P. Zhang, X.Z. Dong, et al., JR6, a new compound isolated from Justicia procumbens, induces apoptosis in human bladder cancer EJ cells through caspasedependent pathway, J. Ethnopharmacol. 144 (2012) 284-292. |
| [4] | F. Qiu, S.J. Zhou, S.J. Fu, et al., LC-ESI-MS/MS analysis and pharmacokinetics of 6'-hydroxy justicidin A, a potential antitumor active component isolated from Justicia procumbens, in rats, J. Pharm. Biomed. 70 (2012) 539-543. |
| [5] | P. Zhang, W.Q. Zhou, X.Z. Dong, M.H. Yang, M.G. Bi, Effect of 6'-hydroxy justicidin A on cell proliferation and redox system in tumor cells, Chin. J. Pharmacol. Toxicol. 24 (2010) 207-213. |
| [6] | P.Zhang, X.Z.Dong, Y.T.Zhou,M.H.Yang,M.G. Bi,Mechanismof JR6 to inducehuman bladder cancer EJ cells apoptosis, Chin. Pharmacol. Bull. 25 (2009) 173-174. |
| [7] | J.Y. Deng, M.H. Yang, M.G. Bi, In vitro anti-tumor mechanism of JR6 on human colon cancer cell HT-29, Chin. Pharmacol. Bull. 25 (2009) 190-191. |
| [8] | (a) P. Allevi, M. Anastasia, P. Ciuffreda, E. Bigatti, Stereoselective glucosidation of podophyllum lignans. A new simple synthesis of etoposide, J. Org. Chem. 58 (1993) 4175-4178; (b) M.G. Saulnier, D.R. Langley, J.F. Kadow, Synthesis of etoposide phosphate, BMY-40481: a water-soluble clinically active prodrug of etoposide, Bioorg. Med. Chem. Lett. 4 (1994) 2567-2572. |
| [9] | L. Xiong, M.G. Bi, S. Wu, Y.F. Tong, Total synthesis of 6'-hydroxy justicidin A, J. Asian Nat. Prod. Res. 14 (2012) 322-326. |
| [10] | (a) S.P. Wang, Y.F. Tong, L. Li, S. Wu, Y. Qi, The synthesis of justicidinoside B and its atropisomer, J. Asian Nat. Prod. Res. 15 (2013) 644-649; (b) J.L. Charlton, C.J. Oleschuk, G.L. Chee, Hindered rotation in arylnaphthalene lignans, J. Org. Chem. 61 (1996) 3452-3457. |
| [11] | M.C. Carreno, J.L. Garcia Ruano, G. Sanz, M.A. Toledo, A. Urbano, N-bromosuccinimide in acetonitrile: a mild and regiospecific nuclear brominating reagent for methoxybenzenes and naphthalenes, J. Org. Chem. 60 (1995) 5328-5331. |
| [12] | W.G. Zhang, J.G. Lin, X.S. Yao, et al., Total synthesis of two new dihydrostilbenes from Bulbophyllum odoratissimum, J. Asian Nat. Prod. Res. 9 (2007) 23-28. |
| [13] | Data of compounds 3 and 8. Compound 3: White solid, mp 95-96℃. 1H NMR (300 MHz, DMSO-d6): δ 3.74 (s, 3H, -OCH3), 3.77 (s, 3H, -OCH3), 3.98-3.89 (m, 2H, C-CH2-C), 4.14-4.03 (m, 2H, C-CH2-C), 5.82 (s, 1H, Ar-CH-O), 7.03 (s, 1H, Ar-H), 7.12 (s, 1H, Ar-H). 13C NMR (100 MHz, DMSO-d6):δ190.8, 155.2, 149.3, 126.4, 120.0, 116.6, 111.1, 65.5, 63.5, 57.1, 56.3. HRESIMS Calcd. for C11H14BrO4 [M+H]+: 289.0070, found: 289.0064. Compound 8: White solid, mp 66-68℃. 1H NMR (400 MHz, DMSO-d6):δ5.23 (s, 2H, Ar-CH2-O), 6.14 (s, 2H, O-CH2-O), 7.09 (s, 1H, Ar-H), 7.61-7.19 (m, 6H, Ar-H×6), 9.75 (s, 1H, -CHO). 13CNMR(100 MHz, DMSOd6):δ191.5, 149.9, 143.2, 141.6, 137.0, 132.1, 129.2, 128.8, 128.6, 113.6, 103.4, 103.1, 71.3. HRESIMS Calcd. for C15H13O4 [M+H]+: 257.0808, found: 257.0799. |
| [14] | Data of compounds 9-14. Compound 9: White solid, mp 115-116℃. 1H NMR (400 MHz, DMSO-d6):δ3.66 (s, 3H, -OCH3), 3.72 (s, 3H, -OCH3), 3.97-3.84 (m, 2H, C-CH2-C), 4.11-3.98 (m, 2H, C-CH2-C), 5.10 (s, 2H, Ar-CH2-O), 5.75 (s, 1H, Ar-CH-O), 5.90 (s, 1H, Ar-CH-Ar), 5.92 (s, 2H, O-CH2-O), 6.47 (s, 1H, Ar-H), 6.70 (s, 1H, Ar-H), 6.92 (s, 1H, Ar-H), 6.97 (s, 1H, Ar-H), 7.48-7.21 (m, 5H, Ar-H×5). 13C NMR (125 MHz, DMSO-d6):δ149.7, 148.8, 147.9, 142.1, 140.5, 137.4, 137.1, 134.2, 128.8, 128.4, 128.2, 126.7, 111.0, 109.9, 108.3, 101.5, 101.2, 100.5, 71.0, 69.3, 65.1, 56.1, 56.0. HRESIMS Calcd. for C26H26NaO8 [M+Na]+: 489.1520, found: 489.1512. Compound 10: White solid,mp 162-164℃. 1H NMR (400 MHz, DMSOd6):δ0.94 (t, 3H, J=7.1 Hz, -CH3), 1.25 (t, 3H, J=7.1 Hz, -CH3), 3.60 (s, 3H-OCH3), 3.93 (s, 3H, -OCH3), 3.95 (q, 2H, J=7.1 Hz, C-CH2-C), 4.33 (q, 3H, J=7.1 Hz, C-CH2-C), 5.14 (s, 2H, Ar-CH2-O), 6.06 (s, 1H, O-CH-O), 6.09 (s, 1H, O-CH-O), 6.45 (s, 1H, Ar-H), 6.56 (s, 1H, Ar-H), 6.71 (s, 1H, Ar-H), 7.48-7.27 (m, 5H, Ar-H×5), 7.63 (s, 1H, Ar-H), 11.92 (s, 1H, Ar-OH). 13C NMR (125 MHz, DMSO-d6):δ169.0, 168.6, 157.3, 152.2, 151.5, 149.1, 148.1, 141.6, 136.7, 134.5, 131.4, 130.7, 129.4, 128.4, 127.9, 127.6, 111.7, 105.6, 104.7, 103.2, 102.6, 101.4, 76.9, 70.4, 55.5, 55.2, 52.3, 51.6, 13.6, 13.5. HRESIMS Calcd. for C32H31O10 [M+H]+: 575.1912, found: 575.1902. Compound 11: White solid, mp 206-207℃. 1H NMR (300 MHz, DMSO-d6):δ3.58 (s, 3H, -OCH3), 3.92 (s, 3H, -OCH3), 5.13 (s, 2H, Ar-CH2-O), 5.36 (s, 2H, Ar-CH2-O), 6.09 (s, 2H, O-CH2-O), 6.53 (s, 1H, Ar-H), 6.62 (s, 1H, Ar-H), 6.90 (s, 1H, Ar-H), 7.46-7.24 (m, 5H, Ar-H×5), 7.62 (s, 1H, Ar-H), 10.50 (s, 1H, Ar-OH). 13C NMR (150 MHz, DMSO-d6):δ169.5, 150.5, 149.7, 148.1, 144.9, 141.7, 136.8, 134.7, 129.4, 129.4, 129.3, 128.4, 127.9, 127.7, 123.2, 121.7, 118.7, 111.4, 105.5, 105.0, 101.3, 100.7, 70.4, 66.6, 55.5, 55.1. HRESIMS Calcd. for C28H23O8 |
| [15] | Compound 15: White solid, mp 212-213℃. The HRESIMS of 15 established a molecular formula of C22H19O11P [m/z 513.0642 (M+Na)+, Calcd. for C22H19NaO11P: 513.0640], indicating 14 degrees of unsaturation. 1H NMR spectrum showed three methoxy groups [δH 3.70 (s, 3H, H-13), 3.94 (s, 3H, H-14), 4.13 (s, 3H, H-15)] and four aromatic protons [δH 7.51 (s, 1H, H-5), 7.05 (s, 1H, H-8), 6.70 (s, 1H, H-20), 6.83 (s, 1H, H-60)]. 13C NMR and DEPT spectra exhibited 22 carbons, including three methyls, two methylenes, sixteen quaternary carbons and one carbonyl. 31P NMR spectrum revealed the existence of one phosphate ester. The HMBC correlations of H-5/C-4, C-7, C-9 and H-8/C-1, C-6, C-10 revealed the present of naphthyl (ring A and ring B). The correlations from H-12 to C-2, C-3, C-4 and C-11 suggested the present of ring C. The correlations of H-13/C-7, H-14/ C-6 and H-15/C-4 gave the substituted positions of three methoxy groups. The linkage of ring B and D was elucidated by the HMBC correlations from H-20 and H-60 to C-1 and C-10. The correlations from H-70 to C-40 and C-50 revealed the presence of ring E. The planar structure of 15 was thus established. 1H NMR (400 MHz, DMSO-d6):δ7.51 (s, 1H, H-5), 7.05 (s, 1H, H-8), 5.71 (s, 2H, H-12), 3.70 (s, 3H, H-13), 3.94 (s, 3H, H-14), 4.13 (s, 3H, H-15), 6.70 (s, 1H, H-20), 6.83 (s, 1H, H-60), 6.13 (d, 2H, J=4.1 Hz, H-70). 13C NMR (125 MHz, DMSO-d6):δ132.3 (C-1), 116.9 (C-2), 119.4 (C-3), 147.8 (C-4), 101.3 (C-5), 151.7 (C-6), 150.5 (C-7), 106.0 (C-8), 128.6 (C-9), 129.7 (C-1'), 169.4 (C-11), 67.3 (C-12), 55.7 (C-13), 56.1 (C-14), 59.6 (C-15), 125.3 (C-1'), 107.6 (C-2'), 148.9 (C-3'), 137.2 (C-4'), 148.9 (C-5'), 116.9 (C-6'), 102.1 (C-7'). 31P NMR (200 MHz, DMSO-d6):δ5.83. |
| [16] | National Pharmacopoeia Committee, Pharmacopoeia of People’s Republic of China. Part 2, vol. XIV, Chemical Industry Press, Beijing, 2010. |