



 , Firoz A. Kalam Khana, Rashmi S. Chouthea, Manoj G. Damaleb, Devanand B. Shindec
, Firoz A. Kalam Khana, Rashmi S. Chouthea, Manoj G. Damaleb, Devanand B. Shindec
b Department of Bioinformatics, MGM Institute of Biosciences and Technology, Aurangabad 431003, India;
c Department of Chemical Technology, Dr. Babasaheb Ambedkar Marathwada University, Aurangabad 431004, India
In recent years,the incidence of systemic fungal infection is increasing dramatically due to an increase in number of patients undergoing organ transplants,anticancer chemotherapy and patients with AIDS. Commonly used antifungal drugs are azoles (fluconazole,itraconazole,miconazole and voriconazole),polyenes (amphotericin B and nystatin),echinocandins (caspofungin and micafungin) and allylamines (naftifine and terbinafine) [1]. Azoles have broad-spectrum activities against most yeasts and filamentous fungi and are the drug of choice for antifungal chemotherapy [2]. Azoles,especially triazole,such as fluconazole,voriconazole and itraconazole,are leading drugs used for the treatment of invasive fungal infections. These antifungal drugs act by inhibiting CYP51 in the process of biosynthesis of ergosterols through a mechanism in which the heterocyclic nitrogen atom (N-4 of triazole) binds to the heme iron atom [3]. However,increasing use of these antifungal drugs has led to increase in resistance to these drugs [4, 5, 6].
The sulfur and nitrogen containing heterocycles,such as thiazole and its derivatives like thienopyridine and tetrahydrothienopyridine, have attracted continuing interest because of their varied biological activities [7, 8, 9, 10]. The triazole ring,difluorophenyl group and hydroxyl group are the pharmacophores of antifungal agents,but the side chains located in the narrow hydrophobic cleft are also important [11, 12, 13, 14]. The optimization of these side chains attached to the pharmacophores has attracted current researches for development of new antifungal agents. In this research work, we intended to alter the side chains to find potent and broad spectrum antifungal agents. The most commonly used method for the preparation of 1,2,4-triazole involves dehydrative condensation of hydrazide derivatives; other approaches involve the Pinner reaction and the Pellizzari reaction. However,these conventional methods involve high reaction temperature,long reaction times and also result in low yields of product [15].
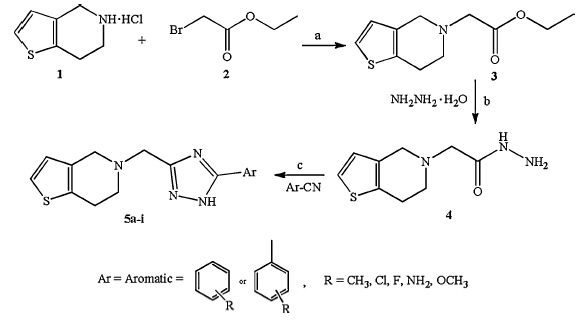
Based on the above facts and our interest for search for new antifungal agents [16, 17, 18, 19, 20, 21],we have reported a facile method for the synthesis of novel,5-((5-substituted-1H-1,2,4-triazol-3-yl)methyl)-4,5,6,7-tetrahydrothienopyridines 5a-i as antifungal agents by using 4-dimethylaminopyridine (DMAP) as a catalyst under microwave irradiation. In present study,we have also reported molecular docking and ADMET properties of the synthesized compounds. The results suggest that the compounds could be exploited as an antifungal drug. 2. Experimental 2.1. Chemistry 2.1.1. Synthesis of 2-(6,7-dihydrothienopyridin-5(4H)-yl)acetohydrazide (4)
Ethyl-2(6,7-dihydrothienopyridine-5(4H)-yl)acetate (3) was obtained by the reaction of 4,5,6,7-tetrahydrothieno[3,2- c]pyridine hydrochloride (1) with ethyl-2-bromoacetate (2) using triethylamine as catalyst [22]. Equimolar quantities of ethyl-2(6,7- dihydrothienopyridine-5(4H)-yl)acetate (3) was refluxed with hydrazine hydrate in n-butanol using glacial acetic acid as catalyst to give product 2-(6,7-dihydrothienopyridin-5(4H)- yl)acetohydrazide (4) [23]. The structures were confirmed by spectral analysis (Mass,1H NMR and 13C NMR) and in agreement with published data. 2.1.2. General procedure for the synthesis of 5-((5-substituted-1H- 1,2,4-triazol-3-yl)methyl)-4,5,6,7-tetrahydrothienopyridines 5a-i
In an Erlenmeyer flask,2-(6,7-dihydrothienopyridin- 5(4H)-yl)acetohydrazide (4) (1.0 mmol),aromatic nitriles (1.0 mmol) and DMAP (30 mol%) were taken in ethanol (15 mL). The reaction mixture was irradiated inside a synthetic microwave oven (Make-Ethosi Milestone with temperature control) for about 20-25 min (700 W). After completion of reaction (monitored by TLC),the mixture was concentrated to obtain the solid product. The solid product formed was filtered,dried and recrystallized from ethanol. 2.2. In vitro antifungal activity
The synthesized compounds 5a-i were screened for their in vitro antifungal activity. The antifungal activity was evaluated against five human pathogenic fungal strains,such as Candida albicans (NCIM3471),Fusarium oxysporum (NCIM1332),Aspergillus flavus (NCIM539),Aspergillus niger (NCIM1196),Cryptococcus neoformans (NCIM576),which are often encountered clinically, and were compared with standard drugs like fluconazole and miconazole. Minimum inhibitory concentration (MIC) values were determined using standard agar method [24]. 2.3. Computational studies 2.3.1. Docking study
The 3D model structure of cytochrome P450 lanosterol 14ademethylase of C. albicans was built using homology modeling with the help of VLifeMDS 4.3 ProModel. Amino acid sequence of enzyme was obtained from the Universal Protein Resource (http:// www.uniprot.org/) (Accession Code: P10613) and sequence homologous was obtained from Protein Data Bank (PDB) using Blast search. Based on the result of blast search,we used the crystal structure of human lanosterol 14a-demethylase (CYP51) with azole as a template for homology modeling (PDB ID: 3LD6). The alignment of amino acid sequence of CA-CYP51 (P10613) and human CYP51 (3LD6_B) is given in Fig. S1 (Supporting information). The quality of the generated C. albicans lanosterol 14ademethylase model was assessed by using the well-validated program like PROCHECK [25] and its structural validation is shown in Fig. S2 (Supporting information). The molecular docking study of the synthesized compounds 5a-i was performed against homology built cytochrome P450 lanosterol 14α-demethylase of C. albicans to understand the binding interactions using VLife MDS 4.3 package following standard procedures [26]. 2.3.2. ADMET properties
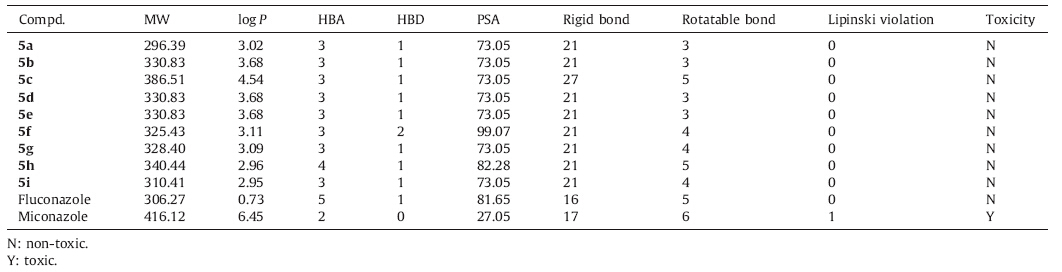
A computational study of synthesized compounds 5a-i was performed for prediction of ADMET properties. In this study,we assessed ADMET properties using ADMET predictor FAFDrugs2 which runs on Linux OS. This tool is freely available and used for in silico ADMET filtering [27]. In particular,we calculated the compliance of synthesized compounds to the Lipinski’s rule of five [28]. This approach has been widely used as a filter for substances that would likely be further developed for drug design programs. We have also assessed parameters like number of rotatable bonds (>10) and the number of rigid bonds which signify that the compound may have good oral bioavailability and good intestinal absorption [29]. 3. Results and discussion 3.1. Chemistry
The synthetic protocols employed for the synthesis of 5-((5-substituted-1H-1,2,4-triazol-3-yl)methyl)-4,5,6,7-tetrahydrothieno[ 3,2-c]pyridine derivatives 5a-i are presented in Scheme 1. For the synthesis of 5-((5-substituted-1H-1,2,4-triazol-3-yl) - methyl)-4,5,6,7-tetrahydrothienopyridine derivatives 5a-i, we first optimized the reaction conditions. The reaction of 2-(6,7- dihydrothienopyridin-5(4H)-yl)acetohydrazide (1.0 mmol) (4) and benzonitrile (1.0 mmol) in ethanol (15 mL) under microwave irradiation (700 W) was used as the model reaction. The catalysts (30 mol%) like K2CO3,triethylamine and 4-dimethylaminopyridine (DMAP) were used to test their efficacy for the synthesis of model reaction. Among the result,a good yield was obtained by the use of catalyst DMAP (91% yield) (Table S1, Supporting information). After deciding upon the catalyst DMAP, the effect of catalyst load was studied at various loads like 10,20, 30 and 40 mol%. DMAP with 30 mol% gave good yield (91%) (Table S2,Supporting information). The effect of various solvents like acetonotrile,isopropylalcohol,n-butanol and ethanol were also studied and ethanol was found to be best among the studied solvents (Table S3,Supporting information). The synthetic protocol was extended for the synthesis of 5-((5-substituted-1H-1,2,4- triazol-3-yl)methyl)-4,5,6,7-tetrahydrothienopyridines 5a- i using various substituted aromatic nitrites under microwave irradiation (700 W) in ethanol with DMAP (30 mol%) as catalyst for about 20-25 min. The purity of the synthesized compounds was checked by TLC and melting points were determined in open capillary tubes melting point apparatus and are uncorrected. The physical data of the synthesized compounds are presented in Table 1. The data obtained from 1H NMR,13C NMR and Mass Spectrometry confirmed the proposed structures (Spectral data results are provided in Supporting information). The products were obtained in good yield (88%-91%) and required less reaction time (20-25 min).
| Table 1 Physical data for 5-((5-substituted-1H-1,2,4-triazol-3-yl)methyl)-4,5,6,7 tetrahydrothienopyridine derivatives 5a-i.a |

The results of in vitro antifungal activity (Table 2) showed that all the compounds had good to moderate antifungal activity. All the synthesized compounds 5a-i were less active against C. albicans,F. oxysporm,A. flavus,A. niger and C. neoformans when compared with fluconazole. Among the synthesized compounds, compound 5g showed significant activity against C. albicans,A.flavus and A. niger when compared with fluconazole. The compound 5g (MIC = 15 μg/mL) showed higher activity against C. albicans than miconazole (MIC = 25 μg/mL). The compounds 5b (MIC = 25 μg/mL against C. albicans) and 5g (MIC = 25 and 12.5 μg/mL against F. oxysporm and A. niger,respectively) were equipotent to miconazole (MIC = 25,25 and 12.5 μg/mL against C. albicans,F. oxysporm and A. niger,respectively). The compounds 5g (MIC = 15 μg/mL against A. flavus) and 5h (MIC = 15 μg/mL against A. niger) showed moderate activitywhen comparedwith miconazole (MIC = 12.5 and 12.5 μg/mL against A. flavus and A. niger,respectively). The compounds 5a and 5i against A. flavus,5a,5c and 5i against A. niger and 5c and 5i against C. neoformans showed no activity up to the concentration of 200 μg/mL.

|
Download:
|
| Scheme 1.Synthetic route for target compounds 5a-i. Reagents and conditions: (a) Acetonitrile,TEA,reflux,5 h. (b) n-butanol,glacial acetic acid,reflux,15 h. (c) DMAP, ethanol,microwave irradiation (700 W),20-25 min. | |
From the antifungal activity data in Table 2,it is observed that scaffolds 1,2,4-triazole and 4,5,6,7-tetrahydrothienopyridine are responsible for antifungal activity. The unsubstituted phenyl analog 5a shows significant activity against C. albicans and F. oxysporum than against C. neoforman. Substituted phenyl analogs are more active than unsubstituted phenyl analogs against almost all of the tested organisms,except for compounds 5c and 5i where the activity is reduced due to substitution. Introduction of p-Cl to phenyl 5b increases the antifungal activity against all of the tested organisms compared to unsubstituted analog 5a. Replacement of p-Cl with o-Cl (5e) and m-Cl (5d) decreases the activity. Introduction of -NH2 at 2-position of phenyl 5f shows moderate activity against C. albicans and F. oxysporum and shows a decrease in activity against A. flavus,A. niger,C. neoformans compared to the unsubstituted analog 5a. Introduction of 4-F to benzyl ring 5g leads to a potent compound against all the tested fungal strains. Compound 5h with 4-OH to benzyl ring shows enhanced activity against A. niger,A. flavus and F. oxysporum as compare to unsubstituted benzyl derivative 5i.
| Table 2 Antifungal activity and results of docking study of compounds. |

3.3.1. Docking study
The synthesized compounds 5a-i and standard drug (fluconazole) were docked into the active site of cytochrome P450 lanosterol 14α-demethylase of C. albicans using VLifeMDS 4.3 software package to understand the binding interactions. The data obtained from docking study is presented in Table 3. The docking results indicated that thienopyridine core of these compounds 5a-i held in the active pocket by forming the hydrophobic interactions with amino acid residues ALA343,THR347,LEU406, LEU412,SER414,MET415,VAL440,SER441,PRO442,PHE499, GLY500,HIS504,ARG505,CYS506,GLY508,ALA512,GLU509, CYS506,LEU412,and ALA501. The compounds 5g and 5i had shown hydrogen bonding interaction with amino acid residues GLU509,CYS506 and LEU412,ALA501,respectively. The halogen substituted compounds 5b,5d,5e and 5g were more favorable for hydrophobic interactions as compare to other substituted (H,-NH2,and OCH3) compounds. The 4-F substituent at benzyl ring of most active compound 5g fitted well into the hydrophobic pocket. The binding interactions for compound 5g and fluconazole are shown in Fig. 1. On the basis of activity data and docking result, it was found that compound 5g had potential to inhibit cytochrome P450 lanosterol 14α-demethylase of C. albicans.

|
Download:
|
| Fig. 1.Docking of compound 5g (Upper left panel),Fluconazole (Upper right panel). Ligands are shown in red color. Hydrogen bonds are shown in green color. Hydrophobic bonds are shown in sky blue color. | |
We had analyzed various physical descriptors and pharmaceutically relevant properties for ADMET prediction by using FAFDrugs2 and data is summarized in Table 3. All the compounds showed significant values for the various parameters analyzed and showed good drug-like characteristics based on Lipinski’s rule of five. The data obtained for all the synthesized compounds 5a-i was within the range of accepted values. None of the synthesized compounds had violated the Lipinski’s rule of five. The value of polar surface area (PSA) for synthesized compounds 5a-i indicated good oral bioavailability. The parameters,like number of rotatable bonds and number of rigid bonds are linked with intestinal absorption result,showed that all synthesized compounds 5a-i had good absorption. All the synthesized compounds were found to be nontoxic.
| Table 3 Prediction of ADMET properties of compounds. |
{kind=link}
{kind=link}
In conclusion,synthesis and antifungal activity of a novel series of 5-((5-substituted-1H-1,2,4-triazol-3-yl)methyl)-4,5,6,7-tetrahydrothieno[ 3,2-c]pyridine derivatives 5a-i has been presented. Use of DMAP as catalyst with ethanol under microwave irradiation helped in fast conversion of 2-(6,7-dihydrothieno pyridine- 5(4H)-yl)acetohydrazide to 5-substituted-1,2,4-triazole derivatives in good yields,proving its advantage. Based on the activity data,structure-activity relationship (SAR) for the series has been developed and it is observed that compound 5b was equipotent with miconazole against C. albicans,whereas compound 5g was equipotent with miconazole against F. oxysporum and A. niger and more potent than miconazole against C. albicans. Also compounds 5d,5e,5f and 5h showed antifungal activity comparable to miconazole against C. albicans,F. oxysporum,A. flavus and A. niger. The structure activity relationship study has suggested that the compound from the present series with 4-Cl or 4-F substituent on phenyl group on 5-position and 4,5,6,7-tetrahydrothienopyridene on 3-position of 1,2,4-triazole can serve as an important gateway for the design and development of new antifungal agent with potent activity. The docking studies of synthesized compounds with lanosterol 14α-demethylase (CYP51) modeled protein showed good binding interactions and formed various hydrophobic interactions with active site residues. Furthermore,analysis of the ADMET parameters for newly prepared compounds showed good drug like properties and opens the possibility for further optimization of studied compounds. Acknowledgments
The authors are thankful to the Mrs. Fatima Rafiq Zakaria Chairman Maulana Azad Educational Trust and Dr. Zahid Zaheer, Incharge Principal,Y.B. Chavan College of Pharmacy,Dr. Rafiq Zakaria Campus,Aurangabad 431 001 (M.S.),India for providing the laboratory facility. Appendix A. Supplementary data
Supplementary data associated with this article can be found,in the online version,at http://dx.doi.org/10.1016/j.cclet.2014.04.003.
| [1] | D.J. Sheehan, C.A. Hitchcock, C.M. Sibley, Current and emerging azole antifungal agents, Clin. Microbiol. Rev. 12 (1999) 40-79. |
| [2] | R. Cha, J.D. Sobel, Fluconazole for the treatment of candidiasis: 15 years experience, Expert Rev. Anti. Infect. Ther. 2 (2004) 357-366. |
| [3] | N.H. Georgopapadakou, T.J. Walsh, Antifungal agents: chemotherapeutic targets and immunologic strategies, Antimicrob. Agents Chemother. 40 (1996) 279-291. |
| [4] | M.A. Pfaller, S.A. Messer, R.J. Hollis, et al., In vitro activities of posaconazole (Sch 56592) compared with those of itraconazole and fluconazole against 3685 clinical isolates of Candida spp. and Cryptococcus neoformans, Antimicrob. Agents Chemother. 45 (2001) 2862-2864. |
| [5] | L. Jeu, F.J. Piacenti, A.G. Lyakhovetskiy, H.B. Fung, Voriconazole, Clin. Ther. 25 (2003) 1321-1381. |
| [6] | G.I. Lepesheva, N.G. Zaitseva, W.D. Nes, et al., CYP51 from Trypanosoma cruzi: a phyla-specific residue in the B0 helix defines substrate preferences of sterol 14ademethylase, J. Biol. Chem. 281 (2006) 3577-3585. |
| [7] | K. Liaras, J. Geronikaki, J. Glamočlija, A.Ćirić,M. Soković, Thiazole-based chalcones as potent antimicrobial agents: synthesis and biological evaluation, Bioorg. Med. Chem. 19 (2011) 3135-3140. |
| [8] | R.G. Kalkhambkar, G.M. Kulkarni, H. Shivkumar, R.R. Nagendra, Synthesis of novel triheterocyclic thiazoles as anti-inflammatory and analgesic agents, Eur. J. Med. Chem. 42 (2007) 1272-1276. |
| [9] | A.B. Scheiff, S.G. Yerande, A. El-Tayeb, et al., 2-Amino-5-benzoyl-4-phenylthiazoles: development of potent and selective adenosine A1 receptor antagonists, Bioorg. Med. Chem. 18 (2010) 2195-2203. |
| [10] | Y. Jiang, J. Zhang, Y. Cao, et al., Synthesis, in vitro evaluation and molecular docking studies of new triazole derivatives as antifungal agents, Bioorg. Med. Chem. Lett. 21 (2011) 4471-4475. |
| [11] | X. Chai, J. Zhang, Y. Cao, et al., Design, synthesis and molecular docking studies of novel triazole as antifungal agent, Eur. J. Med. Chem. 46 (2011) 3167-3176. |
| [12] | P.H. Olesen, A.R. Sorensen, B. Urso, et al., Synthesis and in vitro characterization of 1-(4-aminofurazan-3-yl)-5-dialkylaminomethyl-1H-[1,2,3]triazole-4-carboxylic acid derivatives: a new class of selective GSK-3 inhibitors, J. Med. Chem. 46 (2003) 3333-3341. |
| [13] | H.J. Breslin, T.A. Miskowski, M.J. Kukla, et al., Tripeptidyl-peptidase II (TPP II) inhibitory activity of (S)-2,3-dihydro-2-(1H-imidazol-2-yl)-1H-indoles, a systematic SAR evaluation. Part 2, Bioorg. Med. Chem. Lett. 13 (2003) 4467-4471. |
| [14] | J.J. Baldwin, P.A. Kasinger, F.C. Novello, et al., 4-Trifluoromethylimidazoles and 5-(4-pyridyl)-1,2,4-triazoles, new classes of xanthine oxidase inhibitors, J. Med. Chem. 18 (1975) 895-900. |
| [15] | Y. Kap-Sun, E. Michelle, J.F. Farkas, N.A. Meanwell, A base-catalyzed, direct synthesis of 3,5-disubstituted 1,2,4-triazoles from nitriles and hydrazides, Tetrahedron Lett. 46 (2005) 3429-3432. |
| [16] | J.N. Sangshetti, A.R. Chabukswar, D.B. Shinde, et al., Microwave assisted one pot synthesis of some novel 2,5-disubstituted 1,3,4-oxadiazoles as antifungal agents, Bioorg. Med. Chem. Lett. 21 (2011) 444-448. |
| [17] | J.N. Sangshetti, D.B. Shinde, One pot synthesis and SAR of some novel 3-substituted 5,6-diphenyl-1,2,4-triazines as antifungal agents, Bioorg. Med. Chem. Lett. 20 (2010) 742-745. |
| [18] | J.N. Sangshetti, R.R. Nagawade, D.B. Shinde, Synthesis of novel 3-(1-(1-substituted piperidin-4-yl)-1H-1,2,3-triazol-4-yl)-1,2,4-oxadiazol-5(4H)-one as antifungal agents, Bioorg. Med. Chem. Lett. 19 (2009) 3564-3567. |
| [19] | J.N. Sangshetti, D.B. Shinde, Synthesis and SAR of some new 4-substituted 3H-1,2,3,5-oxathiadiazole 2-oxides as antifungal agents, Lett. Drug Des. Discov. 7 (2010) 171-175. |
| [20] | J.N. Sangshetti, D.B. Shinde, Synthesis of some novel 3-(1-(1-substitutedpiperidin-4-yl)-1H-1,2,3-triazol-4-yl)-5-substituted phenyl-1,2,4-oxadiazoles as antifungal agents, Eur. J. Med. Chem. 46 (2011) 1040-1044. |
| [21] | J.N. Sangshetti, D.B. Shinde, A.P. Sarkate, Synthesis, antifungal activity and docking study of some new 1,2,4-triazole analogs, Chem. Biol. Drug Des. 78 (2011) 800-809. |
| [22] | V.P. Modi, P.N. Patel, H.S. Patel, Synthesis, spectral investigation and biological evaluation of novel noncytotoxic tetrahydrothieno [3,2-c]pyridine hydrazide derivatives, Der Pharmacia Lett. 3 (2011) 120-133. |
| [23] | J.N. Sangshetti, P.D. Priyanka, S.C. Rashmi, et al., Microwave assisted nano (ZnO-TiO2) catalyzed synthesis of some new 4,5,6,7-tetrahydro-6-((5-substituted-1,3,4-oxadiazol-2-yl) methyl) thieno[2,3-c] pyridine as antimicrobial agents, Bioorg. Med. Chem. Lett. 23 (2013) 2250-2253. |
| [24] | D. Greenwood, R.C.B. Slack, J.F. Peutherer, Medical Microbiology, 4th ed., ELBS, London, 1992. |
| [25] | R.W. Hooft, G. Vriend, C. Sander, E.E. Abola, Errors in protein structures, Nature 381 (1996) 272. |
| [26] | VLife Molecular Design Suite 4.3, VLife Sciences Technologies Pvt. Ltd., www.Vlifesciences. com. |
| [27] | D. Lagorce, H. Sperandio, M.A. Miteva, et al., FAF-Drugs2: free ADME/tox filtering tool to assist drug discovery and chemical biology projects, BMC Bioinformatics 9 (2008) 396. |
| [28] | C.A. Lipinski, F. Lombardo, B.W. Dominy, et al., Experimental and computational approaches to estimate solubility and permeability in drug discovery and development settings, Adv. Drug Deliv. Rev. 46 (2001) 3-26. |
| [29] | P. Ertl, B. Rohde, P. Selzer, Fast calculation of molecular polar surface area as a sum of fragment-based contributions and its application to the prediction of drug transport properties, J. Med. Chem. 43 (2000) 3714-3717. |