



 , Cheng Jianga,b,c
, Cheng Jianga,b,c
b Department of Medicinal Chemistry, School of Pharmacy, China Pharmaceutical University, Nanjing 210009, China;
c Jiangsu Key Laboratory of Carcinogenesis and Intervention, China Pharmaceutical University, Nanjing 210009, China;
d Gansu Institute for Food and Drug Control, Lanzhou 730070, China
DNA topoisomerase I (Top I) is a ubiquitous enzyme that resolves superhelical tension and other topological consequences that occur during separation of two DNA strands. Top I relaxes DNA supercoil by relieving torsional stress due to various DNA metabolic processes,including replication,transcription,recombination,chromatin condensation and chromosome partitioning in cell division. In accordance,topoisomerase activities are increased in cancer cell proliferation,thus topoisomerases are proven to be effective targets for antineoplastic drugs [1, 2].
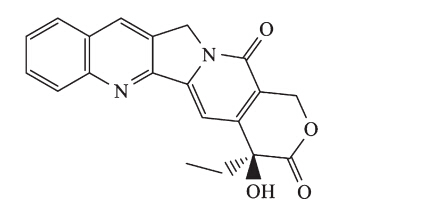
Camptothecin (CPT,Fig. 1) and its analogs are specific inhibitors of Top I and widely used in clinics. However,the therapeutic potential of CPT and its derivatives is thwarted by their rapid inactivation through lactone ring hydrolysis at physiological pH. Therefore,some metabolically stable non-CPT derivatives have been developed [3,45, 6, 7,8],such as indolocarbazoles [7] and indenoisoquinolines [8],some of which are now in clinical evaluation. It could be reasoned that better chemical stability with longer lifetimes of the trapped cleavage complex of the non-CPT Top I inhibitors was due to the absence of a lactone ring in their skeleton [9]. The newly discovered templates retain some of the characteristics of the original template but exhibit improved potency,selectivity,and pharmacokinetics profiles.

|
Download:
|
| Fig. 1. The structure of camptothecin (CPT). | |
Our group has previously identified a series of quinolone and naphthyridine derivatives as novel Top I inhibitors by using a scaffold modification strategy [10]. In continuation of this work, we herein present the synthesis and biological studies of a series of 3-substituted pridopyrimidine compounds as potential anticancer agents by inhibiting Top I. 2. Experimental
2.1. Chemistry
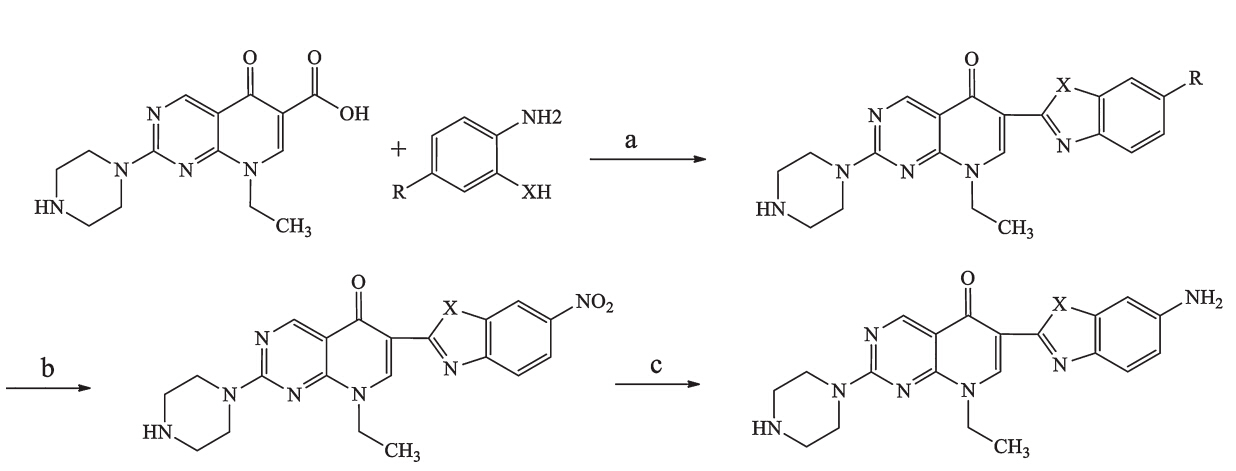
The target compounds 1-13 (Table 1) were synthesized as outlined in Scheme 1. Pipemidic acid was condensed with o-phenylenediamine,o-aminophenol,oro-aminobenzenethiol in polyphosphoric acid (PPA) to obtain 1-7. Subsequently,1-3 were subjected to nitration in a mixture of concentrated sulfuric acid and nitric acid (1:1.2-1.3) to afford 8-10. Hydrogenation of 8 and 9 gave 11 and 12. Compound 10 was reduced using iron powder to afford 13. Details of synthesis and spectral data are provided in Supporting information.
| Table 1 Structures andin vitro MTT assay results for target compounds. |

{kind=link}

|
Download:
|
| Scheme 1. Synthesis of target compounds. Reagents and conditions: (a) PPA,N2,170-250 °C,9.7%-40.9%; (b) HNO3,H2SO4,40-45 °C,30.5%-54.8%; (c) Pd/C,H2or Fe,H3O+, 30.5%-44.8%. | |
{kind=link}
We used the 1-N-methyl-5-thiotetrazole (MTT)-based assay to evaluate the antiproliferative effect of all 14 compounds in four cancer cell lines,A549,BGC-823,SMMC-7721,and HL-60. Cell suspension was made of 2-104/mL to 4-104/mL. The cells were seeded in 96-well tissue plates and treated for 24 h in thermostat CO2incubator. Tested compounds were added and treated for 48 h. Twenty microlitres of MTT solution were added to each well,and plates were incubated for 4 h. DMSO (150mL) was added to each well and mixed thoroughly to dissolve the dark blue crystals. The absorbance was measured on an enzyme-linked immunosorbent detector using a tested wavelength of 570 nm. 2.3. Top I inhibition assays
Top I activity was measured by the relaxation of superhelical DNA in plasmid pBR322. Each 20mL assay contained 5mL Tris-HCl (pH 8.1) which including 100 mmol/L KCl,5 mmol/L DTT and 100 mg/L BSA,pBR322 DNA (0.25mg),and tested compounds at various concentrations. The reaction was started by the addition of 1 U of Top I and allowed to proceed at 37°C for 30 min. 2uL stop solution containing 10% SDS,0.5% bromophenol blue,and 50% glycerol was added to stop the reaction. The solution was subjected to electrophoresis in 1% agarose gel at 15 V for 12 h in 1-TBE buffer. The gels were stained with 1.0 mg/L ethidium bromide for 30 min. The DNA bands were visualized by illumination from below with UV 260 nm and photographed [11, 12]. 2.4. Docking study
Glide was selected as the molecular docking tool. The crystallized complex structure of Top1 (PDB ID: 1K4T) was prepared using the Protein PreparationWizard workflow. A receptor grid was generated on the center of the cocrystallized ligand,which was defined as the ligand-binding site search region. The compounds to be docked were confirmed by an enclosing box that was similar in size to the cocrystallized ligand. Furthermore, the compound set was minimized using the LigPrep module. The best conformation of each compound was output on the basis of the Glide score and interactions formed between the compounds and the active site. Finally,the potential compounds were flexibly docked into the binding site using the extra precision (XP) docking mode. All the remaining parameters were kept as default. 3. Results and discussion
We used the 1-N-methyl-5-thiotetrazole (MTT)-based assay to evaluate the antiproliferative effect of the target compounds in four cancer cell lines: A549,HL60,BGC-823,and SMMC-7721. CPT was used as the positive control. Table 1 lists cytotoxic activities of pridopyrimidine derivatives containing three kinds of heterocycles,such as benzimidazole,benzoxazole,and benzothiazole at the 3-position. By comparing the basic skeletons of these three heterocycles in 1-3,the most active structural feature relevant to cytotoxicity against the four tumor cell lines appeared to be the 3-benzothiazole substituted pridopyrimidine (3) with an IC50in 1.05-3.27 umol/L range. Most of the compounds with an electronwithdrawing group at the 6-position of the benzimidazole, benzoxazole,and benzothiazole rings (5,7,8,9,10) exhibited better cytotoxicity (IC50range 0.13-8.02mmol/L) than 1-3. Among these compounds,9,pridopyrimidine with a 3-benzoxazole substituent,had an IC50range of 0.13-0.84mmol/L,better than the other compounds containing a 3-benzimidazole or 3-benzothiazole ring. Compound 9 show comparable cytotoxic activities to CPT. Compound 10,a pridopyrimidine with a 3-benzothiazole substituent,had little change in cytotoxic activity compared to3. However,11-13,obtained by further reduction of the nitro group of 8-10,had dramatically reduced cytotoxicity. This result lent supported our presumption that an electronwithdrawing group at position 6 was critical for cytotoxicity.
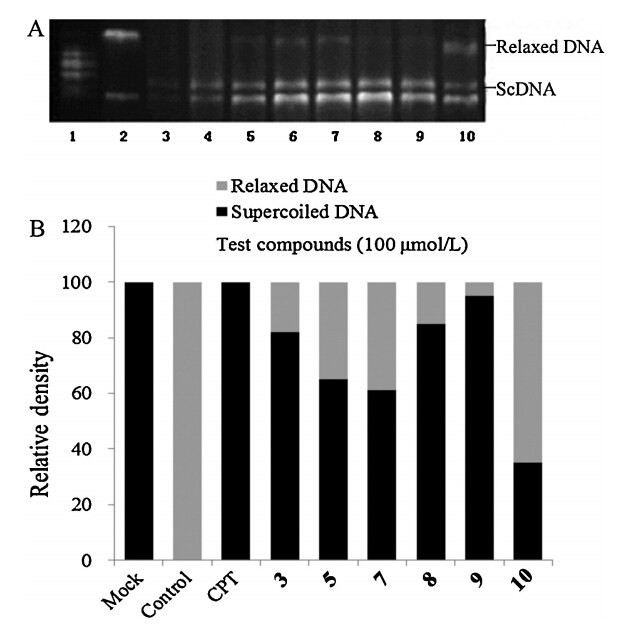
In order to investigate the mechanism of action if our compounds inhibited Top I and thereby caused cytotoxicity,six compounds with the best cytotoxic activity (3,5,7,8,9,10) were examined at 100mmol/L by measuring the relaxation of supercoiled DNA of plasmid pBR322. We also compared the inhibitory activities of these this of derivatives with a well-known DNA-Top I inhibitor,CPT. Results were shown in Fig. 2. The data showed that all tested compounds had moderate inhibitory activities on Top I, which was in coincidence with the results of cytotoxicity activity. Compound 9 showed best activity,which was as potent as CPT.

|
Download:
|
| Fig. 2. Effects of eight compounds on Top I-mediated DNA relaxation. (A) The position of the origin and the migration positions of relaxed and supercoiled plasmids. Lane 1: markers; Lane 2: Top I + ScDNA + DMSO; Lane 3: supercoiled pBR322 DNA (ScDNA); Lane 4: Top I + ScDNA + CPT; Lane 5: Top I + ScDNA +3; Lane 6: Top I + ScDNA +5; Lane 7: Top I + ScDNA + 7; Lane 8: Top I + ScDNA +8; Lane 9: Top I + ScDNA +9; Lane 10: Top I + ScDNA +10. (B) Effects of test compounds on Top I-mediated DNA relaxation. The proportion of relaxed DNA to total DNA was measured by scanning with an imaging system. Bars show the percentage of supercoiledvsrelaxed DNA. | |
{kind=link}
In order to know the binding mode of 9 with Top I/DNA binary complex,a docking study was performed and the result was shown as Fig. 3. It could be find that the rigid plane formed by the pridopyrimidine core and 3-benzoxazole substituent inserted in to Top I/DNA binary complex,and an H-bond was formed between the oxygen atom at C-4 position and Arg364 of Top I. Usually,to our knowledge,the piperazine could enhance the water solubility of compounds. Interestingly,in our docking result,the NH of the piperazine group at C-7 position of 9 formed an H-bond with Glu356. This may improve the binding affinity of the inhibitor with Top I/DNA binary complex.

|
Download:
|
| Fig. 3. Docking result of9with Top1/DNA binary complex. | |
{kind=link}
In summary,a series of new pyridopyrimidine derivatives were designed and evaluated as novel Top I inhibitors using a scaffold modification strategy. Many of these compounds were effective against tested cancer cells. All the tested compounds showed moderate inhibitory activities on Top I,which was in coincidence with the results of cytotoxic activity assays. Among them, compound 9 showed the best activity which was as potent as CPT. Lead optimization and other molecular mechanism of action studies are currently under way in our lab. This discovery may yield novel lead compounds for further modification/optimization in the development of potent Top I inhibitors as anti-cancer agents. Acknowledgments
This work was supported by the National Natural Science Foundation of China (No. 21072232),the Fundamental Research Funds for the Central Universities (No. JKZ2011002),the National Science & Technology Major Project (Nos. 2012ZX09304-001 and 2013ZX09103-001-007),the Program for Changjiang Scholars and Innovative Research Team in University (No. PCSIRT-IRT1193) and the ‘‘Qinglan Project’’ of Jiangsu Province. Appendix A. Supplementary data
Supplementary material related to this article can be found,in the online version,at http://dx.doi.org/10.1016/j.cclet.2014.05.048.
| [1] | L.F. Liu, DNA topoisomerase poisons as antitumor drugs, Annu. Rev. Biochem. 58 (1989) 351-375. |
| [2] | B.K. Sinha, Topoisomerase inhibitors. A review of their therapeutic potential in cancer, Drugs 49 (1995) 11-19. |
| [3] | B.M. Fox, X. Xiao, S. Antony, et al., Design, synthesis, and biological evaluation of cytotoxic 11-alkenylindenoisoquinoline topoisomerase I inhibitors and indenoisoquinoline-camptothecin hybrids, J. Med. Chem. 46 (2003) 3275-3282. |
| [4] | A. Morrell, S. Antony, G. Kohlhagen, et al., A systematic study of nitrated indenoisoquinolines reveals a potent topoisomerase I inhibitor, J. Med. Chem. 49 (2006) 7740-7753. |
| [5] | Y. Pommier, Topoisomerase I inhibitors: camptothecins and beyond, Nat. Rev. Cancer 6 (2006) 789-802. |
| [6] | X.S. Xiao, S. Antony, Y. Pommier, et al., Total synthesis and biological evaluation of 22-hydroxyacuminatine, J. Med. Chem. 49 (2006) 1408-1412. |
| [7] | Z.Y. Li, F.M. Zhai, L. Zhao, et al., Design and synthesis of N-methylmaleimide indolocarbazole bearing modified 2-acetamino acid moieties as Topoisomerase I inhibitors, Bioorg. Med. Chem. Lett. 19 (2009) 406-409. |
| [8] | X.Y. Zhang, R. Wang, L. Zhao, et al., Synthesis and biological evaluations of novel indenoisoquinolinesas topoisomerase I inhibitors, Bioorg. Med. Chem. Lett. 22 (2012) 1276-1281. |
| [9] | Y. Pommier, DNA topoisomerase I inhibitors: chemistry, biology, and interfacial inhibition, Chem. Rev. 109 (2009) 2894-2902. |
| [10] | Q.D. You, Z.Y. Li, C.H. Huang, et al., Discovery of a novel series of quinolone and naphthyridine derivatives as potential topoisomerase I inhibitors by scaffold modification, J. Med. Chem. 52 (2009) 5649-5661. |
| [11] | M.M. Bradford, A rapid and sensitive method for the quantitation of microgram quantities of protein utilizing the principle of protein-dye binding, Anal. Biochem. 72 (1976) 248-254. |
| [12] | E. Aflalo, S. Iftach, S. Segal, et al., Inhibition of topoisomerase I activity by tyrphostin derivatives, protein tyrosine kinase blockers: mechanism of action, Cancer Res. 54 (1994) 5138-5142. |