



 , Qin-Sheng Sunb, Lu-Lu Lib, Chun-Yan Tanb,c, Hong-Xia Liub, Yu-Yang Jiangb,c,d
, Qin-Sheng Sunb, Lu-Lu Lib, Chun-Yan Tanb,c, Hong-Xia Liub, Yu-Yang Jiangb,c,d
b The Ministry-Province Jointly Constructed Base for State Key Lab-Shenzhen Key Laboratory of Chemical Biology, Shenzhen 518055, China;
c Shenzhen Anti-Tumor Drug Development Engineering Laboratory, the Graduate School at Shenzhen, Tsinghua University, Shenzhen 518055, China;
d School of Medicine, Tsinghua University, Beijing 100084, China
Cancer is one of the most common malignant diseases obsessing mankind and new effective drugs are needed [1, 2, 3, 4, 5, 6]. DNA topoisomerase,either in prokaryotes or eukaryotes,plays a very important role in cell proliferation,survival and apoptosis, which has been one of the most potential targets for the development of new anticancer agents [7, 8, 9].
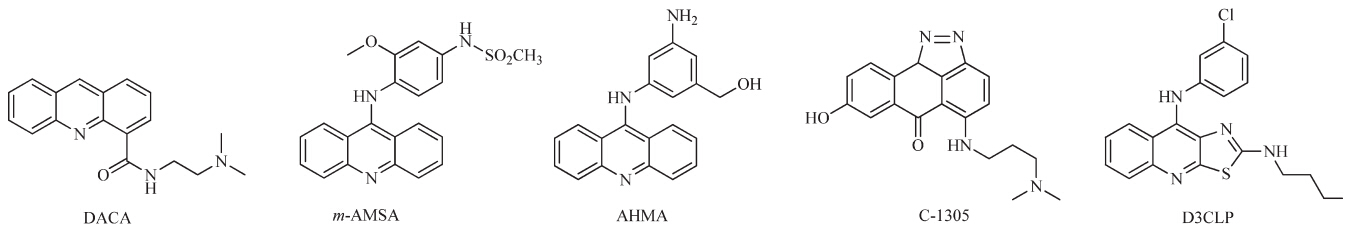
Acridine analogs have been used for treatment of inflammation and cancer for many years. The unique planar ring structure makes its strong interaction with DNA base pairs [10, 11]. A variety of acridine derivatives have been designed and synthesized,some of which have entered clinical studies,such as DACA [12, 13, 14],C-1305 [15] andm-AMSA,etc.(Fig. 1) [16]. Among them,m-AMSA was the first used in clinical treatment for several cancers as topoisomerase inhibitors and much attention had been paid to the modification of m-AMSA to improve its activity and bioavailability [17]. A variety of analogs ofm-AMSA,such as AHMA and D3CLP (Fig. 1),have been developed [18, 14]. By far,most of the modification of them-AMSA was focused on the position and nature of substituents in the 9-aminobenzene moiety and acridine rings. A variety of bis-acridine derivatives have also been developed to increase the DNA binding affinity. However,little attention has been paid to the replacement of benzene ring by naphthalene or heterocycles,such as pyridine.

|
Download:
|
| Fig. 1. The structures of some acridine derivatives. | |
Previously,we have found that the linker between acridine and benzene ring played an important role in the antitumor activity and identified the -NHCH2- or -NHCH2CH2- linker was appropriate [18] as part of our continuous efforts [18, 16, 19, 20, 21, 22, 23] in developing novel antitumor compounds. In this paper we synthesized a series of naphthalene,pyridine and indole substituted-acridine analogs with -NHCH2- or -NHCH2CH2- linker as potent antitumor agents. Our results indicated that some of these 9-heteroaromatic substituted acridines displayed better antiproliferative activity than the corresponding 9-benzene substituted acridine 5b[18]. The DNA binding capability and topoisomerase I (topo I) mediated relaxation of plasmid pBR322 DNA were also evaluated. 2. Experimental
The synthetic methods and the preparation of compounds 3 and 4 can be found in the Supporting information.
General procedure for compounds (7a-7d): Initially,compound 4(1.0 mmol) and phenol (10 mmol) were added to a 100 mL dried round-bottom. The mixture was incubated at 60°C for 1 h under argon atmosphere to give the intermediate5. Then,the corresponding amine 6(1.1 mmol) was added and the mixture was heated to 130°C for 2 h. The mixture was then poured into a mixture ofN-methyl morpholine (1 mL) and ethyl acetate (100 mL). The precipitation was separated by vacuum filtration to give the crude products (7a-7d) (Scheme 1).

|
Download:
|
| Scheme 1. Synthesis of acridine derivatives 7a-7d. Reagents and conditions: (i) K2CO3,Cu,DMF,130 °C; (ii) POCl3,105 °C; (iii) phenol,60°C. (iv) the corresponding amines 7a-7d,120 °C. | |
N-(2-(1H-indol-3-yl)ethyl)acridin-9-amine (7a). Compound 7a was purified by column chromatography (methanol/ethyl acetate = 1/4,v/v). Yield 57%; mp 245-247°C; 1H NMR (400 MHz, DMSO-d6):δ 10.92 (s,1H),8.60 (d,2H,J= 8.3 Hz),8.00-7.86 (m,4H), 7.58 (d,1H,J= 7.8 Hz),7.50 (t,2H,J= 6.7 Hz),7.33 (d,1H,J= 8.1 Hz), 7.25 (d,1H,J= 2.2 Hz),7.06 (t,1H,J= 7.2 Hz),6.95 (t,1H,J= 7.2 Hz), 5.30-5.29 (m,1H),4.37 (t,2H,J= 7.4 Hz),4.35 (t,2H,J= 7.4 Hz). HRMS (ESI): Calcd. for [M+H]+ :m/z338.1657; found: 338.1670.
N-((Pyridin-4-yl)methyl)acridin-9-amine (7b). Compound 7b was purified by recrystallization from ethyl acetate. Yield 27%; mp 225-228°C; 1H NMR (400 MHz,DMSO-d6): δ8.75 (s,1H),8.67- 8.48 (m,3H),8.03-7.95 (m,4H),7.92 (d,1H,J= 7.8 Hz),7.51 (s,2H), 7.43 (dd,1H,J= 7.7,4.8 Hz),5.41 (s,2H). HR-MS (ESI): Calcd. for [M+H]+:m/z286.1344; found: 286.1345.
N-((Pyridin-2-yl)methyl)acridin-9-amine (7c). Compound 7c was purified by recrystallization from ethyl acetate. Yield 25%; mp 228-229°C; 1H NMR (400 MHz,DMSO-d6): δ8.77 (s,1H),8.66- 8.46 (m,3H),8.08-7.97 (m,2H),7.92-7.74 (m,3H),7.52-7.45 (m, 3H),5.41 (s,2H). HR-MS (ESI): Calcd. for [M+H]+: m/z286.1344; found: 286.1338.
N-((Naphthalen-1-yl)methyl)acridin-9-amine (7d). Compound 7d was purified by recrystallization from DMSO/ethyl acetate (1/1, v/v). Yield 46%; mp 249-252°C; 1H NMR (400 MHz,DMSO-d6):δ 8.13-8.09 (m,2H),8.09-8.04 (m,1H),8.01-7.93 (m,5H),7.73 (d, 1H,J= 7.0 Hz),7.65 (dd,3H,J= 6.3,3.3 Hz),7.59-7.51 (m,1H),7.43 (s,2H),5.77 (s,2H). HR-MS (ESI): Calcd. for [M+H]+:m/z335.1548; found: 335.1533.
The experiments of UV-vis absorption spectroscopy and fluorescence emission; DNA topo I inhibition assay; 1H NMR and high resolution mass spectra can be found in Supporting information. 3. Results and discussion
Synthesis of the acridine-based derivatives 7a-7d was accomplished as described in Scheme 1. First,an Ullmann coupling reaction of 2-chloro-benzoic acid1with aniline 2 in DMF using Cu as the catalyst gave anthranilic acid 3,which was then stirred in POCl3 to afford the 9-chloroacridine 4. The reaction of 4 with phenol gave the intermediate5,which was then reacted with the corresponding amines to afford the desired acridines7a-7d.
MTT assay was used to test whether compounds7a-7d displayed antiproliferative activity. The cytotoxicity of compounds 7a-7d against K562 and HepG-2 cells was evaluated for comparison with that of the 9-benzylamino acridine (5b) reported in our earlier paper [18]. Colchicine and podophyllotoxin were used as the positive controls. The results can be reported in Table 1. The newly synthesized four acridines displayed moderate to good antiproliferative activity against the two tumor cell lines. The 4-pyridine substituted acridine7bshowed about 3-fold less activity against K562 cells than 5b. The other three compounds demonstrated similar,or more cytotoxicity compared to 5b. The low micromolar IC50 values of the compounds indicate that the replacement of benzene ring by naphthalene or other heterocycles may improve the antitumor activity. As the mode of toxic action of acridines is mainly attributed to DNA and its related enzymes, compound 7c with the best antiproliferative activity against K562 cells was selected to study the DNA binding property.
| Table 1 Antiproliferative activity of compounds against K562 and HepG-2 cells. |

{kind=link}
{kind=link}
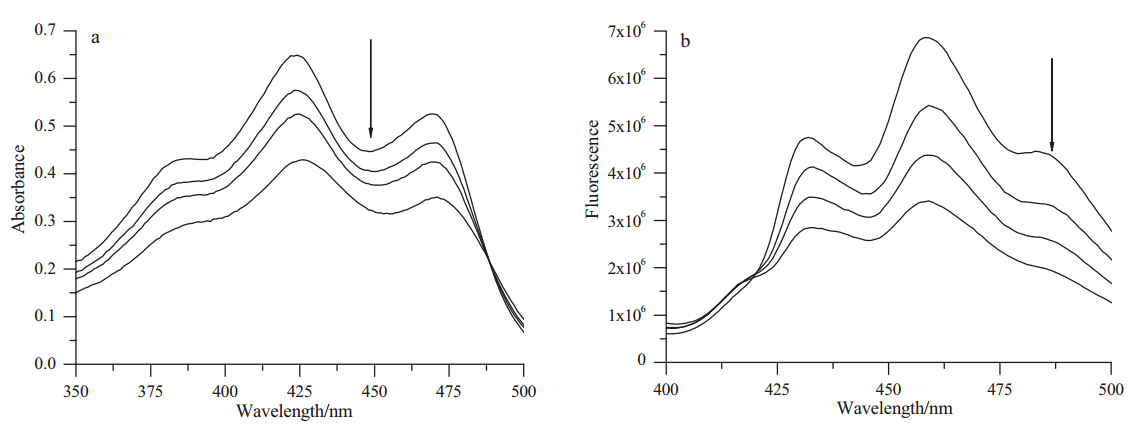
UV-vis absorption spectroscopy is extensively utilized to detecting the interaction between DNA and compounds. The interaction of compound 7c with ctDNA was evaluated and the result shown in Fig. 2a. The major peaks in the spectrum of 7c were observed at about 426 nm and 470 nm,while DNA did not absorb light in this region. The absorption spectra of 7c decreased obviously by the addition of increasing amounts of ctDNA. In addition,a slight bathochromic effect was observed when ctDNA was added and a clear isosbestic point appeared. All the result suggested that compound 7c can interact with DNA.

|
Download:
|
| Fig. 2. (a) UV-vis absorption spectra of 7c(62.5mmol/L) in 10 mmol/L Tris-HCl buffer containing 10 mmol/L NaCl (pH 7.0) by increasing the concentrations of ctDNA ([DNA]/ [7c] = 0,0.125,0.25,0.5). The arrow indicates the absorbance changes upon increasing DNA concentrations. (b) Spectrofluorimetric titration of 7c (62.5mmol/L) in the presence of increasing amounts of ctDNA ([DNA]/[7c] = 0,1,2,4). The arrow indicates the fluorescence emission changes upon increasing DNA concentrations. | |
{kind=link}
In addition to UV-vis absorption spectroscopy,fluorescence emission spectroscopy is also widely used to evaluate the binding ability of drugs and their targets. As shown inFig. 2b,the fluorescence of 7c was gradually decreased when the concentration of ct DNA increased,which suggested that there are interactions between 7c and ct DNA.
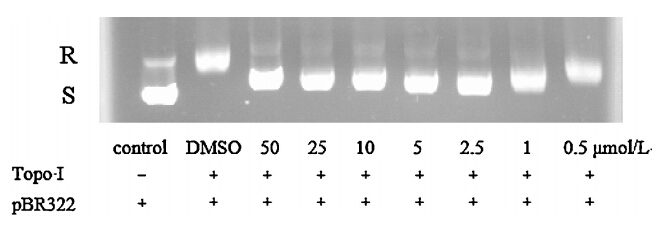
Compound 7c can interact with DNA,which will cause distortion of the DNA structure. Therefore,by the activity of topo I,the DNA related enzyme will be inhibited. To identify whether compound7ccould inhibit topo I,the assay of compound7con the relaxation of plasmid pBR322 DNA mediated by topo I was performed. As seen from Fig. 3,compound7cdisplayed excellent topo I inhibitory activity at about 1mmol/L,whereas had undetectable activities at 0.5mmol/L,which was in accordance with its antiproliferative activity. These data suggested that the antitumor activity of7cwas attributed to its DNA binding and topo I inhibition,and might be a potential lead compound to be developed into novel topo I inhibitors.

|
Download:
|
| Fig. 3. Effect of the compound7con the relaxation of plasmid DNA by human topo I (S: superhelix; R: relaxation). | |
{kind=link}
In conclusion,a series of novel 9-aminoacridine derivatives 7a- 7d had been designed and synthesized,most of which showed good antiproliferative activity against both K562 and HepG-2 cells. The identified compound 7c exhibited its antitumor activity through binding with DNA and inhibiting topo I activity,which can be developed into novel topoisomerase inhibitors. Further optimizations of the structure to improve the bioavailability and solubility are ongoing. Acknowledgments
The authors would like to thank the Ministry of Science and Technology of the People’s Republic of China (Nos. 2012AA020305 and 2011DFA30620),the National Natural Science Foundation of China (Nos. 21272134 and 21372141),and Shenzhen Sci. & Tech. Bureau (No. JCYJ20120831165730905) for the financial supports. Appendix A. Supplementary data
Supplementary data associated with this article can be found,in the online version,at http://dx.doi.org/10.1016/j.cclet.2014.03.028.
| [1] | I. Kümler, N. Brünner, J. Stenvang, E. Balslev, D.L. Nielsen, A systematic review on topoisomerase I inhibition in the treatment of metastatic breast cancer, Breast Cancer Res. Treat. 138 (2013) 347-358. |
| [2] | X. Zhang, B. Bao, X.H. Yu, et al., The discovery and optimization of novel dual inhibitors of topoisomerase II and histone deacetylase, Bioorg. Med. Chem. 21 (2013) 6981-6995. |
| [3] | S.M.B. Nijman, S.H. Friend, Cancer. Potential of the synthetic lethality principle, Science 342 (2013) 809-811. |
| [4] | Y. Li, H.B. Zhang, W.L. Huang, X. Zhen, Y.M. Li, Synthesis and biological evaluation of tetrahydroisoquinoline derivatives as potential multidrug resistance reversal agents in cancer, Chin. Chem. Lett. 19 (2008) 169-171. |
| [5] | L.H. Shen, Y. Li, D.H. Zhang, Y.S. Lai, L.J. Liu, Synthesis and evaluation of nitrate derivatives of colchicine as anticancer agents, Chin. Chem. Lett. 22 (2011) 768-770. |
| [6] | C. Kaplan-Ozen, B. Tekiner-Gulbas, E. Foto, et al., Benzothiazole derivatives as human DNA topoisomerase II alpha inhibitors, Med. Chem. Res. 22 (2013) 5798-5808. |
| [7] | Z.Y. Tian, H.X. Ma, S.Q. Xie, et al., Synthesis, DNA binding and topoisomerase inhibition of mononaphthalimide homospermidine derivatives, Chin. Chem. Lett. 19 (2008) 509-512. |
| [8] | J.R. Goodell, A.V. Ougolkov, H. Hiasa, et al., Acridine-based agents with topoisomerase II activity inhibit pancreatic cancer cell proliferation and induce apoptosis, J. Med. Chem. 51 (2012) 179-182. |
| [9] | K. Padget, A. Stewart, P. Charlton, M.J. Tilby, C.A. Austin, An investigation into the formation of N-2-(dimethylamino)ethyl acridine-4-carboxamide (DACA) and 6-2-(dimethylamino)ethylamino-3-hydroxy-7H-indeno[2,1-c]quinolin-7-one dihydrochloride (TAS-103) stabilised DNA topoisomerase I and II cleavable complexes in human leukaemia cells, Biochem. Pharmacol. 60 (2000) 817-821. |
| [10] | J.Y. Chang, C.F. Lin, W.Y. Pan, et al., New analogues of AHMA as potential antitumor agents: synthesis and biological activity, Bioorg. Med. Chem. 11 (2003) 4959-4969. |
| [11] | K. Rastogi, J.Y. Chang, W.Y. Pan, et al., Antitumor AHMA linked to DNA minor groove binding agents: synthesis and biological evaluation, J. Med. Chem. 45 (2002) 4485-4493. |
| [12] | R. Zittoun, m-AMSA: a review of clinical data, Eur. J. Cancer Clin. Oncol. 21 (1985) 649-653. |
| [13] | T.L. Su, T.C. Chou, J.Y. Kim, et al., 9-Substituted acridine-derivatives with long halflife and potent antitumor-activity-synthesis and structure-activity-relationships, J. Med. Chem. 38 (1995) 3226-3235. |
| [14] | I. Gonzalez-Sanchez, J.D. Solano, M.A. Loza-Mejia, et al., Antineoplastic activity of the thiazolo 5, 4-b quinoline derivative D3CLP in K-562 cells is mediated through effector caspases activation, Eur. J. Med. Chem. 46 (2011) 2102-2108. |
| [15] | C.H. Chen, Y.W. Lin, X.G. Zhang, et al., Synthesis and in vitro cytotoxicity of 9-anilinoacridines bearing N-mustard residue on both anilino and acridine rings, Eur. J. Med. Chem. 44 (2009) 3056-3059. |
| [16] | X.D. Luan, C.M. Gao, N.N. Zhang, et al., Exploration of acridine scaffold as a potentially interesting scaffold for discovering novel multi-target VEGFR-2 and Src kinase inhibitors, Bioorg. Med. Chem. 19 (2011) 3312-3319. |
| [17] | Y.Q. Li, C.Y. Tan, C.M. Gao, et al., Discovery of benzimidazole derivatives as novel multi-target EGFR, VEGFR-2 and PDGFR kinase inhibitors, Bioorg. Med. Chem. 19 (2011) 4529-4535. |
| [18] | X.L. Lang, L.L. Li, Y.Z. Chen, et al., Novel synthetic acridine derivatives as potent DNA-binding and apoptosis-inducing antitumor agents, Bioorg. Med. Chem. 21 (2013) 4170-4177. |
| [19] | X.L. Lang, Q.S. Sun, Y.Z. Chen, et al., Novel synthetic 9-benzyloxyacridine analogue as both tyrosine kinase and topoisomerase I inhibitor, Chin. Chem. Lett. 24 (2013) 677-680. |
| [20] | C.M. Gao, F. Liu, X.D. Luan, et al., Novel synthetic 2-amino-10-(3, 5-dimethoxy) benzyl-9(10H)-acridinone derivatives as potent DNA-binding antiproliferative agents, Bioorg. Med. Chem. 18 (2010) 7507-7514. |
| [21] | C.M. Gao, S.F. Li, X.L. Lang, et al., Synthesis and evaluation of 10-(3, 5-dimethoxy) benzyl-9(10H)-acridone derivatives as selective telomeric G-quadruplex DNA ligands, Tetrahedron 68 (2012) 7920-7925. |
| [22] | X.D. Luan, C.M. Gao, Q.S. Sun, et al., Novel synthetic azaacridine analogues as topoisomerase 1 inhibitors, Chem. Lett. 40 (2011) 728-729. |
| [23] | X.L. Lang, X.D. Luan, C.M. Gao, Y.Y. Jiang, Recent progress of acridine derivatives with antitumor activity, Prog. Chem. 24 (2012) 1497-1505. |