




b State Key Laboratory of Biotherapy, West China Hospital, West China Medical School, Sichuan University, Chengdu 610041, China
The fused pyran ring skeleton is a well-known heterocycle and important core unit in a number of natural products [1, 2, 3]. Natural products bearing a fused pyran ring system possess distinct properties of general interest,and have a variety of valuable biological activities and potential medicinal applications [4, 5, 6, 7, 8]. It is worthwhile to note that many of these natural products exhibit cancer cell growth inhibitory activity and have been further investigated as potential anticancer agents [9, 10]. As more valuable properties emerge,these pyran-containing structures begin to inspire chemists to develop efficient synthetic approaches and identify novel biologically active compounds [11, 12].
Recently,multicomponent reactions (MCR) have been used frequently in organic synthesis,and considerable efforts have been focused on the design and development of environmentally friendly and less expensive procedures for the generation of libraries of heterocyclic compounds [13, 14, 15]. Among them,the construction of pyran ring is particularly interesting and valuable, especially those methods that provided a feasible strategy for the production of molecules comprised of two heterocyclic nucleuses [11, 16]. However,these methods are limited by the aldehydes tolerated,which greatly restricted the generation of more diverse pyran analog libraries. Here,we report a mild method for the synthesis of novel 2-amino-4H-pyran derivatives using ethanol as a cheap,safe,and environmentally benign solvent. Their in vitro antitumor activity was evaluated using the National Cancer Institute (NCI) in vitro disease-oriented human cells screening panel assay. 2. Experimental
All reagents were acquired from commercial sources and used without further purification. All reactions were monitored by thin layer chromatography (TLC silica gel 60 F254 plates) analysis. All products were purificated by column chromatography (200-300 mesh silica gel,Qingdao Marine Chemical Ltd.,Qingdao,China). Melting points were measured using an YRT-3 melting point measuring apparatus (Precision Instrument Plant,Tianjin University) and uncorrected. The 1H NMR spectra and 13C NMR spectra were recorded on a Bruker AM400 NMR spectrometer and the chemical shifts in ppm were reported relative to tetramethylsilane (TMS) or residual solvent peaks. Mass spectrometry (ESI-MS) data were mensurated using a Bruker Daltonics amaZon SL mass spectrometer. Crystal data of 4a were collected on a Xcalibur E diffractometer with monochromated Mo Kα radiation (λ = 0.71073A˚ ) at 293 K,and operating in the ω scan mode. The structure was solved with the Superflip structure solution program using Charge Flipping and refined on F2 by the full matrix leastsquares methods using SHELXTL.

|
Download:
|
| Scheme 1.Synthesis of 2-amino-4H-pyran derivatives. | |
General procedures for the synthesis of compounds 4a-4q (Scheme 1): A solution of α,β-unsaturated ketones (1,0.5 mmol), malononitrile (2,0.5 mmol),aldehydes (3,0.5 mmol),DBU (1 mmol) in ethanol (10 mL) was stirred at room temperature for 1 hour or until 1 was completely consumed (monitored by TLC analysis). After the completion of the reactions,ethanol was removed under reduced pressure. The crude products were purified by column chromatography on silica gel,eluting with petroleum ether/ethyl acetate/triethylamine (35:15:1,v/v/v). Further purification of the products was accomplished by recrystallization. Physical and chemical data of chosen products are described below.
(E)-2-Amino-6-methyl-4-phenyl-8-(4-(trifluoromethyl)benzylidene)- 5,6,7,8-tetrahydro-4H-pyrano[3, 2, c]pyridine-3-carbonitrile (4a): White solid; mp 235-238°C; 1H NMR (400 MHz,CDCl3): d 2.26 (s,3H),2.75 (d,1H,J = 16 Hz),2.97 (d,1H,J = 16 Hz),3.36 (d, 1H,J = 13.8 Hz),3.53 (d,1H,J = 13.8 Hz),4.03 (s,1H),4.68 (s,2H), 6.90 (s,1H),7.24-7.39 (m,7H),7.61 (d,2H,J = 8 Hz); 13C NMR (100 MHz,CDCl3):δ41.7,44.9,54.6,55.3,60.5,114.1,119.7,121.4, 122.8,125.3,125.4,125.5,127.7,127.9,128.9,129.0,129.3,129.8, 139.8,139.9,142.0,158.8. MS (ESI): m/z 424.2 [M+H]+. (E)-2-Amino-4-(3-nitrophenyl)-8-(4-(trifluoromethyl)benzylidene)- 5,6,7,8-tetrahydro-4H-chromene-3-carbonitrile (4o): Fulvous solid; mp 211-214°C; 1H NMR (400 MHz,CDCl3):δ1.65- 1.66 (m,2H),1.88-1.92 (m,1H),2.04-2.08 (m,1H),2.54-2.58 (m, 1H),2.69-2.73 (m,1H),4.14 (s,1H),4.69 (s,2H),6.93 (s,1H),7.39- 7.63 (m,6H),8.11-8.17 (m,2H); 13CNMR(100 MHz,CDCl3):δ22.1, 27.0,27.5,43.6,59.4,114.8,119.3,122.3,122.8,125.2,128.7,129.0, 129.4,129.9,130.8,134.2,140.4,141.9,145.0,148.8,159.1. MS (ESI): m/z 454.2 [M+H]+. (E)-tert-Butyl 2-amino-3-cyano-4-(3-nitrophenyl)-8-(4-(trifluoromethyl) benzylidene)-7,8-dihydro-4H-pyrano[3, 2, c]pyridine- 6(5H)-carboxylate (4p): Flavescens solid; mp 210-212°C; 1H NMR (400 MHz,CDCl3):δ1.30 (s,9H),3.63-4.78 (m,7H),6.98 (s, 1H),7.40-7.42 (m,2H),7.57-7.66 (m,4H),8.13-8.20 (m,2H); 13C NMR (100 MHz,CDCl3):δ28.1,40.0,41.3,44.1,59.5,80.9,111.9, 118.8,122.7,123.1,125.5,129.3,130.1,134.1,148.9,153.9,158.9. MS (ESI): m/z 555.3 [M+H]+. (E)-2-Amino-4-(3-nitrophenyl)-8-(4-(trifluoromethyl)benzylidene)- 4,5,7,8-tetrahydropyrano[4, 3, b]pyran-3-carbonitrile (4q):Flavescens solid; mp 238-240°C; 1H NMR (400 MHz,DMSO-d6): d 3.77 (d,1H,J = 16 Hz),4.23 (d,1H,J = 16 Hz),4.44 (s,1H),4.54 (d, 1H,J = 13.8 Hz),4.65 (d,1H,J = 13.8 Hz),7.01 (s,1H),7.12 (s,2H), 7.44-7.46 (m,2H),7.69-7.77 (m,4H),8.09 (s,1H),8.17-8.19 (m, 1H); 13C NMR (100 MHz,DMSO):δ55.3,65.3,65.5,114.1,120.4, 120.9,122.5,123.0,123.3,125.9,128.8,130.2,131.1,134.9,138.9, 139.8,145.7,148.6,160.5. MS (ESI): m/z 456.2 [M+H]+. 3. Results and discussion
As shown in Table 1,we began our optimization studies from the reaction between (E)-1-methyl-3-(4-(trifluoromethyl)benzylidene) piperidin-4-one,malononitrile and benzaldehyde,in equimolecular amounts,using DBU as a base and dichloromethane as a solvent. Under mild conditions at room temperature for 1 h,the domino reaction provided a promising 87% isolated yield of (E)-2- amino-6-methyl-4-phenyl-8-(4-(trifluoromethyl)benzylidene)- 5,6,7,8-tetrahydro-4H-pyrano[3, 2, c]pyridine-3-carbonitrile 4a (entry 1). This model reaction was then performed in acetonitrile and DMF,which led to poorer isolated yields 73% and 72% of 4a, respectively (entries 2-3). To our pleasure,the reaction in ethanol gave an isolated yield of 95% of 4a (entry 6).
| Table 1 Optimization of reaction conditions for compound 4a. |
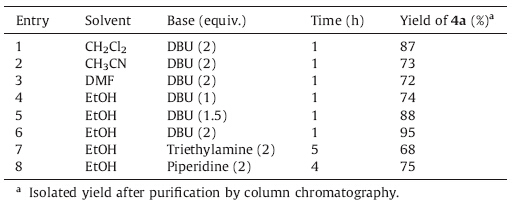
The nature and the amount of base have significant influence on the yield (entries 4-8). It was not advisable to reduce the amount of DBU,which led to a decrease in yields (entries 4-6). In addition,the efficacy of the bases assayed changed in the order: triethylamine < piperidine < DBU (entries 6-8). It was found that the reaction proceeded most efficiently in the presence of 2 equivalents of DBU in ethanol at room temperature for 1 h,and the desired product was isolated in the best yield.
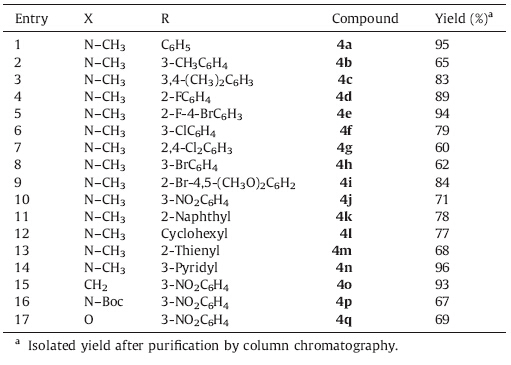
Under the optimized conditions,various a,b-unsaturated ketones (1) with a diverse set of aldehydes (3) were then applied to the preparation of a library of 2-amino-4H-pyran analogs (4). In general,the reactions could proceed smoothly with 60%-96% isolated yields,which were remarkable considering the large number of individual steps involved (Table 2). Firstly,different aromatic aldehydes were applied to the cyclization reaction (entries 1-11). By varying substituent(s) at the benzoyl moiety,we found that the yields varied greatly within an acceptable range. Next,cyclohexanecarboxaldehyde and two heteroaromatic aldehydes were used for the three-component reaction to enrich the diversity of aldehydes (entries 12-14). Finally,(E)-1-methyl-3-(4- (trifluoromethyl)benzylidene)piperidin-4-one was replaced by other α,β-unsaturated ketones to verify the scope of the threecomponent cyclization reaction. As expected,these reactions afforded good yields of desired products (entries 15-17).
| Table 2 Synthesis of 2-amino-4H-pyran derivatives (Scheme 1). |

|
Download:
|
| Scheme 2.The proposed mechanism for the reaction. | |
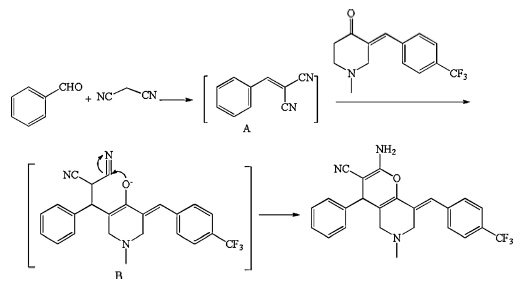
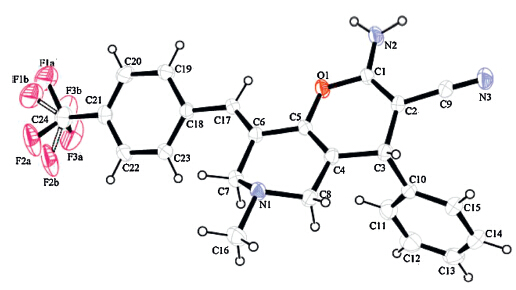
The structures of the targeted compounds 4a-4q were fully characterized by melting points,MS (ESI),1H NMR and 13C NMR. The mass spectrum of compound 4a (Table 2,entry 1) displayed the molecular ion peak at m/z 424.2,which is consistent with the proposed structure. The 1H NMR spectrum of 4a contained singlets for NH2 and N-CH3 at 4.68 and 2.26 ppm,respectively. The geometrical configuration of 4a was confirmed by a single crystal X-ray crystallographic study (Fig. 1) [17]. A proposed mechanism for the reaction is shown in Scheme 2.

|
Download:
|
| Fig. 1.Single crystal structure of compound 4a. | |
The compounds were screened for their in vitro antitumor activity in the full NCI 96 cell panel,including human colon cancer (HCT116),human cervical cancer (Hela),and non-small cell lung cancer (H1975). In the protocol,all compounds were tested initially at 20 μmol/L,and their percentage growth inhibition (GI%) were presented in Table 3. Compounds 4k and 4o exhibited noticeable growth inhibitory activity against the subtotal tested subpanel tumor cell lines,and other compounds also showed some antitumor activity. However,when the concentrations of the test compounds decreased to 10 mmol/L or lower,none of the above compounds showed satisfying activity.
| Table 3 In vitro percentage growth inhibition (GI%) caused by the test compounds at dose of 20μmol/L. |

{kind=link}
{kind=link}
{kind=link}
In summary,an efficient and mild cyclization procedure for the synthesis of novel 2-amino-4H-pyran derivatives using ethanol as a cheap,safe,and environmentally benign solvent and DBU as a base was identified. The present methodology provides a direct and convenient route to construct more diverse pyran analog libraries. The antitumor activity of the synthesized compounds was evaluated in three human tumor cell lines,including human colon cancer (HCT116),human cervical cancer (Hela),and nonsmall cell lung cancer (H1975).
| [1] | N. Sultana, P.G. Waterman, 3-Monoterpenyl-2,4-deoxygenated quinoline alkaloids from the aerial parts of Halfordia kendack, Phytochemistry 58 (2001) 329-332. |
| [2] | K. Nakashima, M. Oyama, T. Ito, et al., Melicodenines A and B, novel Diels-Alder type adducts isolated from Melicope denhamii, Tetrahedron Lett. 52 (2011) 4694-4696. |
| [3] | K. Nakashima, M. Oyama, T. Ito, et al., Novel quinolinone alkaloids bearing a lignoid moiety and related constituents in the leaves of Melicope denhamii, Tetrahedron 68 (2012) 2421-2428. |
| [4] | I.S. Chen, I.W. Tsai, C.M. Teng, et al., Pyranoquinoline alkaloids from Zanthoxylum simulans, Phytochemistry 46 (1997) 525-529. |
| [5] | M. Isaka, M. Tanticharoen, P. Kongsaeree, et al., Structures of cordypyridones A-D, antimalarial N-hydroxy-and N-methoxy-2-pyridones from the insect pathogenic fungus Cordyceps nipponica, J. Org. Chem. 66 (2001) 4803-4808. |
| [6] | C.L. Cantrell, K.K. Schrader, L.K. Mamonov, et al., Isolation and identification of antifungal and antialgal alkaloids from Haplophyllum sieversii, J. Agric. Food Chem. 53 (2005) 7741-7748. |
| [7] | A.U. Rahman, A. Khalid, N. Sultana, et al., New natural cholinesterase inhibiting and calcium channel blocking quinoline alkaloids, J. Enzyme Inhib. Med. Chem. 21 (2006) 703-710. |
| [8] | P.L. Katavic, D.A. Venables, G.P. Guymer, et al., Alkaloids with human delta-opioid receptor binding affinity from the Australian rainforest tree Peripentadenia mearsii, J. Nat. Prod. 70 (2007) 1946-1950. |
| [9] | I.S. Chen, S.J. Wu, I.L. Tsai, et al., Chemical and bioactive constituents from zanthoxylum simulans, J. Nat. Prod. 57 (1994) 1206-1211. |
| [10] | K.D. McBrien, Q. Gao, S. Huang, et al., Fusaricide, a new cytotoxic N-hydroxypyridone from Fusarium sp., J. Nat. Prod. 59 (1996) 1151-1153. |
| [11] | R.R. Kumar, S. Perumal, P. Senthilkumar, P. Yogeeswari, D. Sriram, An atom efficient, solvent-free, green synthesis and antimycobacterial evaluation of 2-amino-6-methyl-4-aryl-8-[(E)-arylmethylidene]-5,6,7,8-tetrahydro-4H-pyrano[3,2-c]pyridine-3-carbonitriles, Bioorg. Med. Chem. Lett. 17 (2007) 6459-6462. |
| [12] | M.N. Erichsen, T.H.V. Huynh, B. Abrahamsen, et al., Structure-activity relationship study of first selective inhibitor of excitatory amino acid transporter subtype 1: 2-amino-4-(4-methoxyphenyl)-7-(naphthalen-1-yl)-5-oxo-5, 6,7,8-tetrahydro-4H-chromene-3-carbonitrile (UCPH-101), J. Med. Chem. 53 (2010) 7180-7191. |
| [13] | R. Doostmohammadi, M.T. Maghsoodlou, N. Hazeri, et al., An efficient one-pot multi-component synthesis of 3,4,5-substituted furan-2(5H)-ones catalyzed by tetra-n-butylammonium bisulfate, Chin. Chem. Lett. 24 (2013) 901-903. |
| [14] | N. Foroughifar, S. Ebrahimi, One-pot synthesis of 1,3-thiazolidin-4-one using Bi(SCH2COOH)3 as catalyst, Chin. Chem. Lett. 24 (2013) 389-391. |
| [15] | R. Baharfar, S.M. Baghbanian, Synthesis of novel uracil based 2,5-diaminofurans using multi-component reactions, Chin. Chem. Lett. 23 (2012) 677-680. |
| [16] | S.L. Wang, Z.G. Han, S.J. Tu, et al., An efficient method for synthesis of pyrano[3,2-c]pyridine derivatives under microwave irradiation, J. Heterocyclic Chem. 46 (2009) 828-831. |
| [17] | Crystallographic data (excluding structure factors) for compound 4a have been deposited with the Cambridge Crystallographic Data Center. |