




b College of Forestry and Horticulture, Hubei Minzu University, Enshi 445000, China
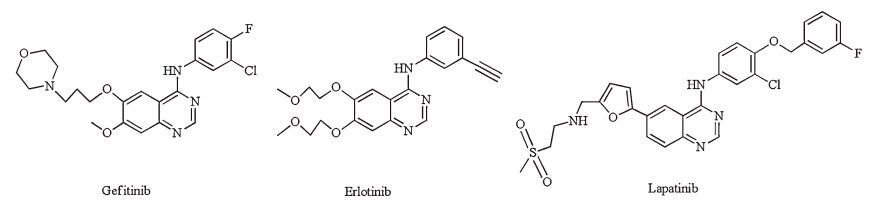
A pyrimidine nucleus fused with another heterocycle is widely used in the design and discovery of novel bioactive molecules and drugs [1]. For example,many anticancer agents that act as tyrosine kinase inhibitors typically contain an aminopyrimidine group as a core moiety. Different quinazoline derivatives,such as gefitinib [2], erlotinib [3] and lapatinib [4] (Fig. 1),have gained market approval worldwide. Recently,the thieno[2, 3, d]pyrimidine core,which is evaluated as a bioisostere of the quinazoline core,was used in the mechanism-based design and synthesis of new antitumor agents [5, 6, 7, 8]. Introducing a fluorine-containing substituent,particularly the trifluoromethyl group,led to higher biological activity and lower toxicity compared with their non-fluorinated analogs [9, 10]. A number of thieno[2, 3, d]pyrimidine derivatives with different substituents at the C-2 and C-4 positions were found to exert potential antitumor activity [11, 12, 13, 14]. However,trifluoromethylsubstituted thieno[2, 3, d]pyrimidines at the C-2 position have seldom been reported [10, 15]. In this work we prepared new thieno[2, 3, d]pyrimidines by introducing the trifluoromethyl group at the C-2 position and different substituents at the C-4 position to explore the potential of thieno[2, 3, d]pyrimidines as antitumor compounds.

|
Download:
|
| Fig. 1.Representative examples of anticancer quinazoline compounds. | |
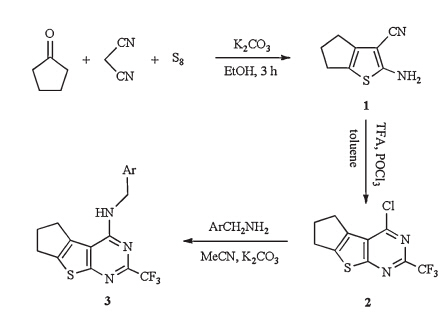
Synthetic protocols for 2,4-disubstituted thieno[2, 3, d]pyrimidines usually involve a conversion of 2-substituted-thieno[2,3- d]pyrimidin-4-ones to 4-chloro-2-substituted-thieno[2, 3, d]pyrimidines, which commonly undergo multi-step procedures (Scheme 1) [16, 17, 18, 19, 20]. These procedures possess disadvantages, such as rigorous conditions,long reaction time,complex handling, and poor total yields. Therefore,developing a facile process to produce 2,4-disubstituted thieno[2, 3, d]pyrimidines is necessary. We utilized a new convenient and efficient method to synthesize a series of novel N4-substituted 2-trifluoromethyl-6,7-dihydro-5Hclopenta[ 4,5]thieno[2, 3, d]pyrimidin-4-amines by reaction of appropriate amines with 4-chloro-2-trifluoromethyl-6,7-dihydro- 5H-clopenta[4, 5]thieno[2, 3, d]pyrimidine,which started directly from 2-amino-5,6-dihydro-4H-cyclopenta[b]thiophene-3-carbonitrile and trifluoroacetic acid (TFA) in the presence of phosphorous oxychloride by a one-pot procedure. Our original synthetic strategy is outlined in Scheme 2.

|
Download:
|
| Scheme 1.The synthetic routes to the NDAZO and PANDAZO. | |

|
Download:
|
| Scheme 2.The synthetic routes to the NDAZO and PANDAZO. | |
All the chemicals used in the synthesis were of analytical grade. IR spectra were recorded on a Nicolet NEXUS 470 FT-IR spectrophotometer in the 4000-400 cm-1 range. NMR spectra were obtained on a Varian XL-400 MHz spectrometer with TMS as the internal standard and DMSO-d6 as the solvent. MS spectra were performed by a Thermo DSQ II mass spectrometer using the electron ionization (EI) method. Elemental analysis was carried out on a Vario EL III CHNSO analyzer,the accepted deviation of experimental values from the calculated ones is 0.3%. X-ray diffraction data were collected on a Bruker Smart APEX-II CCD diffractometer equipped with a graphite-monochromatizedMo Ka (λ = 0.71073A˚ ) radiation. Melting points were measured with an X-4 digital melting-point apparatus and uncorrected.
Synthesis of 2-amino-5,6-dihydro-4H-cyclopenta[b]thiophene- 3-carbonitrile (1): Cyclopentanone (0.84 g,10 mmol),malononitrile (0.66 g,10 mmol),elemental sulfur (0.35 g,11.0 mmol),K2CO3 (0.28 g,2.0 mmol) and 15 mL of dry ethanol were stirred at reflux for 3 h. The insoluble material was filtered off,and the solvent was removed by evaporation under reduced pressure. The crude product was washed with water and recrystallized from ethanol to give yellowish crystals in 81% yield. Mp 152-153°C (Lit. [21]: 151°C).
Synthesis of 4-chloro-2-trifluoromethyl-6,7-dihydro-5H-clopenta[ 4,5]thieno[2, 3, d]pyrimidine (2): A mixture of compound 1 (0.82 g,5 mmol),TFA (0.5 mL),toluene (8 mL) and phosphoryl trichloride (1.5 mL) was heated to 80°C with good stirring. The progress of the reaction was monitored by TLC with petroleum ether-ethyl acetate (3:1,v/v) as a developing solvent. Toluene was removed by vacuum distillation after the completion of the reaction. The residue was poured over crushed ice and neutralized with a saturated sodium bicarbonate solution. The aqueous mixture was extracted with diethyl ether and the organic layer was washed with water followed by saturated aqueous sodium chloride. After evaporation of the solvent,the residue was recrystallized from n-hexane to afford the yellowish compound 2 in 65% yield. Mp 142-143°C. Anal. Calcd. for C10H6ClF3N2S: C 43.10,H 2.17,N 10.05; found: C 42.95,H 2.32,N 9.93.
Synthesis of compounds 3a-3k: A mixture of compound 2 (1.39 g,5 mmol),appropriate amine (5 mmol),and K2CO3 (1.38 g, 10 mmol) in acetonitrile (10 mL) was heated under reflux for 2- 3 h. When the reaction was completed (TLC),acetonitrile was removed by evaporation,the residue was washed with water and purified by normal chromatography to afford the desired products 3a-3k as white solid.
Compound 3a: Yield 86%,mp 128-129°C; 1H NMR (400 MHz, DMSO-d6):δ7.80 (s,1H,NH),7.41-7.20 (m,5H,Ar-H),4.72 (s,2H, Ar-CH2),3.08-2.88 (m,4H,5- and 7-CH2),2.43-2.40 (m,2 H,6- CH2); 19F NMR (376 MHz,DMSO-d6):δ-69.02; IR (KBr,cm-1): 3418 (N-H),1573 (C=N),1344,1126 (CF3); EI-MS (%):m/z 349 (M+, 75.9),106 (78.4),91 (100); Anal. Calcd. for C17H14F3N3S: C 58.44,H 4.04,N 12.03; found: C 58.27,H 3.89,N 11.94.
Compound 3b: Yield 78%,mp 145-146°C; 1H NMR (400 MHz, DMSO-d6):δ7.71 (s,1H,NH),7.33-7.03 (m,4H,Ar-H),4.71 (s,2H, Ar-CH2),3.07-2.90 (m,4H,5- and 7-CH2),2.42-2.39 (m,2 H,6- CH2); 19F NMR (376 MHz,DMSO-d6):δ-69.17,-114.04; IR (KBr, cm-1): 3425 (N-H),1568 (C=N),1361,1137 (CF3); EI-MS (%): m/z 367 (M+,77.6),124 (100),109 (65.2); Anal. Calcd. for C17H13F4N3: C 55.58,H 3.57,N 11.44; found: C 55.70,H 3.46,N 11.52.
Compound 3c: Yield 82%,mp 135-136°C; 1H NMR (400 MHz, DMSO-d6):δ7.83 (s,1H,NH),7.43-7.09 (m,4H,Ar-H),4.67 (s,2H, Ar-CH2),3.09-2.92 (m,4H,5- and 7-CH2),2.50-2.40 (m,2 H,6- CH2); 19F NMR (376 MHz,DMSO-d6):δ-69.00,-116.30; IR (KBr, cm-1): 3469 (N-H),1590 (C=N),1339,1143 (CF3); EI-MS (%): m/z 367 (M+,46.8),124 (58.8),109 (100); Anal. Calcd. for C17H13F4N3S: C 55.58,H 3.57,N 11.44; found: C 55.43,H 3.72,N 11.28.
Compound 3d: Yield 79%,mp 153-154°C; 1H NMR (400 MHz, DMSO-d6):δ7.74 (s,1H,NH),7.43-7.26 (m,4H,Ar-H),4.79 (s,2H, Ar-CH2),3.13-2.95 (m,4H,5- and 7-CH2),2.50-2.45 (m,2H,6- CH2); 19F NMR (376 MHz,DMSO-d6):δ-69.12; IR (KBr,cm-1): 3452 (N-H),1580 (C=N),1367,1121 (CF3); EI-MS (%):m/z 383 (M+, 64.6),140 (61.3),125 (100); Anal. Calcd. for C17H13CIF3N3S: C 53.20,H 3.41,N 10.95; found: C 53.31,H 3.56,N 11.10.
Compound 3e: Yield 84%,mp 126-127°C; 1H NMR (400 MHz, DMSO-d6):δ7.87 (s,1H,NH),7.42-7.31 (m,4H,Ar-H),4.67 (s,2H, Ar-CH2),3.08-2.89 (m,4H,5- and 7-CH2),2.50-2.40 (m,2H,6- CH2); 19F NMR (376 MHz,DMSO-d6):δ-69.01; IR (KBr,cm-1): 3462 (N-H),1568 (C=N),1333,1121 (CF3); EI-MS (%):m/z 383 (M+, 70.0),140 (64.4),125 (100); Anal. Calcd. for C17H13CIF3N3S: C 53.20,H 3.41,N 10.95; found: C 53.40,H 3.27,N 10.86.
Compound 3f: Yield 75%,mp 150-151°C; 1H NMR (400 MHz, DMSO-d6):δ7.72 (s,1H,NH),7.30-7.11 (m,4H,Ar-H),4.68 (s,2H, Ar-CH2),3.13-2.96 (m,4H,5- and 7-CH2),2.50-2.45 (m,2H,6- CH2),2.38 (s,3H,CH3); 19F NMR (376 MHz,DMSO-d6):δ-69.10; IR (KBr,cm-1): 3468 (N-H),1585 (C=N),1339,1121 (CF3); EI-MS (%): m/z 363 (M+,44.0),120 (19.9),105 (100); Anal. Calcd. for C18H16CIF3N3S: C 59.49,H 4.44,N 11.56; found: C 59.64,H 4.29,N 11.46.
Compound 3g: Yield 80%,mp 124-125°C; 1H NMR (400 MHz, DMSO-d6):δ7.67 (s,1H,NH),7.15-6.94 (m,4H,Ar-H),4.52 (s,2H, Ar-CH2),2.97-2.80 (m,4H,5- and 7-CH2),2.37-2.27 (m,2H,6- CH2),2.10 (s,3H,CH3); 19F NMR (376 MHz,DMSO-d6):δ-69.06; IR (KBr,cm-1): 3413 (N-H),1580 (C=N),1334,1137 (CF3); EI-MS (%): m/z 363 (M+,40.6),120 (30.9),105 (100); Anal. Calcd. for C18H16CIF3N3S: C 59.49,H 4.44,N 11.56; found: C 59.37,H 4.53,N 11.75.
Compound 3h: Yield 81%,mp 157-158°C; 1H NMR (400 MHz, DMSO-d6):δ7.89 (s,1H,NH),7.64-7.59 (m,4H,Ar-H),4.77 (s,2H, Ar-CH2),3.11-2.94 (m,4H,5- and 7-CH2),2.49-2.44 (m,2H,6- CH2); 19F NMR (376 MHz,DMSO-d6):δ-61.18,-69.09; IR (KBr, cm-1): 3429 (N-H),1568 (C=N),1322,1126 (CF3); EI-MS (%): m/z 417 (M+,73.6),174 (100),159 (39.2); Anal. Calcd. for C18H13F6N3S: C 51.80,H 3.14,N 10.07; found: C 51.92,H 3.30,N 9.89.
Compound 3i: Yield 74%,mp 154-155°C; 1H NMR (400 MHz, DMSO-d6):δ7.52 (s,1H,NH),7.18-6.84 (m,4H,Ar-H),4.70 (s,2H, Ar-CH2),3.83 (s,3H,OCH3),3.12-2.96 (m,4H,5- and 7-CH2),2.49- 2.44 (m,2H,6-CH2); 19F NMR (376 MHz,DMSO-d6):δ-69.09; IR (KBr,cm-1): 3468 (N-H),1585 (C=N),1361,1121 (CF3); EI-MS (%): m/z 379 (M+,59.5),136 (32.1),121 (100); Anal. Calcd. for C18H16CIF3N3S: C 56.98,H 4.25,N 11.08; found: C 57.15,H 4.09, N 11.23.
Compound 3j: Yield 73%,mp 123-124°C; 1H NMR (400 MHz, DMSO-d6):δ7.70 (s,1H,NH),7.09-6.64 (m,4H,Ar-H),4.55 (s,2H, Ar-CH2),3.57 (s,3H,OCH3),2.98-2.81 (m,4H,5- and 7-CH2),2.37- 2.30 (m,2H,6-CH2); 19F NMR (376 MHz,DMSO-d6):δ-69.06; IR (KBr,cm-1): 3432 (N-H),1579 (C=N),1339,1132 (CF3); EI-MS (%): m/z 379 (M+,68.2),136 (100),121 (57.5); Anal. Calcd. for C18H16CIF3N3OS: C 56.98,H 4.25,N 11.08; found: C 57.11,H 4.34, N 11.20.
Compound 3k: Yield 83%,mp 118-119°C; 1H NMR (400 MHz, DMSO-d6):δ7.77 (s,1H,NH),7.33-6.84 (m,4H,Ar-H),4.62 (s,2H, Ar-CH2),3.68 (s,3H,OCH3),3.07-2.91 (m,4H,5- and 7-CH2),2.49- 2.40 (m,2H,6-CH2); 19F NMR (376 MHz,DMSO-d6):δ- 69.08; IR (KBr,cm-1): 3474 (N-H),1596 (C=N),1367,1115 (CF3); EI-MS (%): m/z 379 (M+,17.0),136 (4.9),121 (100); Anal. Calcd. for C18H16CIF3N3OS: C 56.98,H 4.25,N 11.08; found: C 56.86,H 4.17, N 10.85. 3. Results and discussion
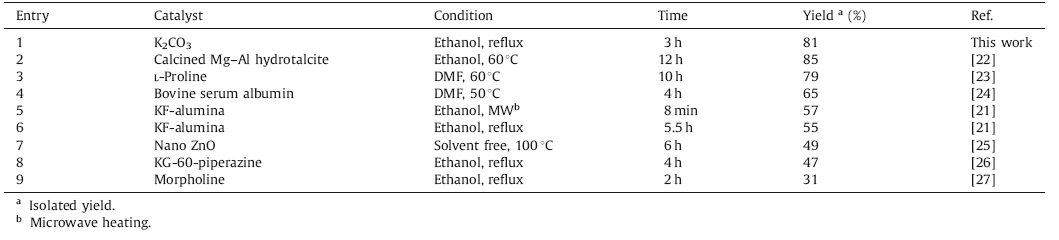
The synthesis was initiated by allowing readily available cyclopentanone to react with malononitrile and sulfur to form thiophene 1 based on the modified Gewald procedure. In this context,we have found that the Gewald reaction efficiently occurs in the presence of potassium carbonate (K2CO3) as a heterogeneous base catalyst under reflux in ethanol. To the best of our knowledge, the use of K2CO3 in the synthesis of 2-aminothiophenes has not been reported. To show the merits of the present work,we compared results obtained from K2CO3 with those previously reported [21, 22, 23, 24, 25, 26, 27]. Table 1 reveals that K2CO3 is an inexpensive, highly efficient,and green catalyst that can produce thiophene 1 in short time and favorable yield. The key intermediate 2 was efficiently prepared directly from thiophene 1,TFA,and phosphorous oxychloride using toluene as a solvent via a one-pot procedure,which presents several advantages,such as milder reaction conditions,simpler handling,and better yields,compared with traditional multi-step methods (Routes A-C in Scheme 1). Subsequently,the chloride 2 reacts with appropriate amines to form 3.
| Table 1 Comparison of different methods for synthesizing thiophene 1 via Gewald reaction of cyclopentanone and malononitrile. |
The structures of compounds 3a-3k were characterized by IR, 1H NMR,19F NMR,EI-MS,and elemental analysis. 1H NMR spectra show the expected occurrence of signals from the NH (δ 7.90- 7.52),aryl protons (δ 7.65-6.60),benzyl CH2 (δ 4.80-4.50),and three cycloalkyl methylene protons (δ 3.15-2.25). The 19F signal assigned to the trifluoromethyl (CF3) group at the C-2 position of the thieno[2, 3, d]pyrimidine ring appears nearδ-69.0. In addition, EI mass spectra gave the anticipated M+ peak. The spectroscopic data are in good agreement with the proposed chemical structures of the synthesized compounds.
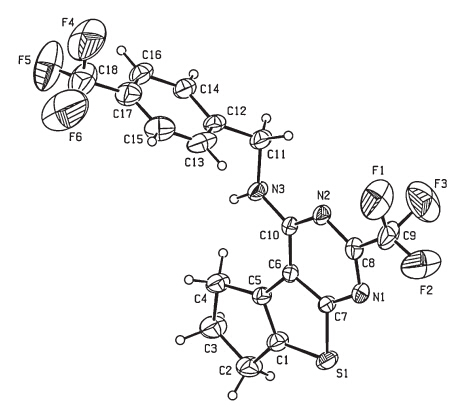
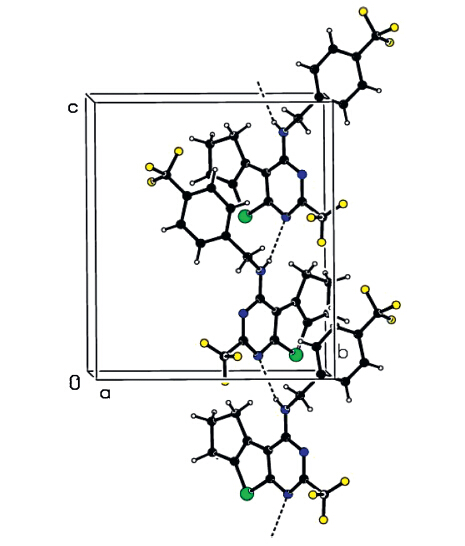
To further confirm the structures of these compounds and provide a basis for the studies of structure-activity relationships, the crystal structure of compound 3h was determined by singlecrystal X-ray diffraction. A colorless single crystal of compound 3h with dimensions of 0.23 mm × 0.21 mm × 0.18 mmwas placed on a Bruker SMART APEX-II diffractometer equipped with a graphitemonochromatized Mo Ka (λ= 0.71073A˚ ) radiation at 298(2) K for analysis. The structure was solved by the direct methods with SHELXS-97 [28] and expanded using the Fourier difference techniques. A total of 9146 reflections were collected in the range of 2.37 ≤ u ≤ 25.258 using a Ψ-ω scan mode with 3294 independent ones (Rint = 0.0397),of which 2134 were observed with I > 2σ(I) and used in the subsequent refinements. The molecular structure and packing diagram are depicted in Figs. 2 and 3, respectively. X-ray diffraction analysis reveals that the thieno[2,3- d]pyrimidine ring,which exhibits good coplanar nature with a maximum deviation of 0.020(6)A˚ at atom C(10),forms a dihedral angle of 75.7(3)° with the benzene ring. The molecular structure is stabilized by intermolecular N(3)-H(3)···(1) hydrogen bonds together with p-p stacking interactions between the benzene and thiophene rings.

|
Download:
|
| Fig. 2.Molecular structure of compound 3h. | |

|
Download:
|
| Fig. 3.Fig. 3. Packing diagram of compound 3h. | |
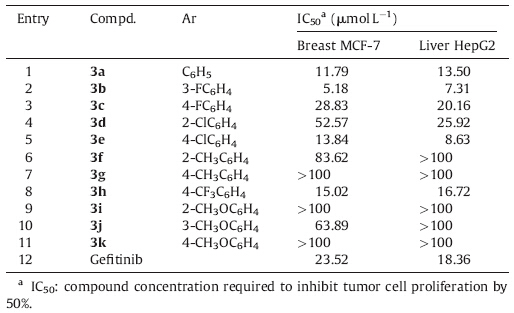
The in vitro antitumor activity of the newly synthesized compounds 3a- 3k against MCF-7 (human breast cancer) and HepG2 (human hepatocellular liver carcinoma) cell lines was evaluated by the standard MTT [3-(4,5-dimethylthiazol-2-yl)-2,5- diphenyl tetrazolium bromide] assay [29] using gefitinib as a positive control. As described in Table 2,the results of preliminary bioassay reveal that compounds 3a,3b,3c,3d,3e and 3h exhibit good antitumor activity against MCF-7 and HepG2. Moreover,3a,3b,3e and 3h possessed higher antitumor activity than the positive control gefitinib. The results imply that different substituents at different positions of the benzene ring significantly affect the antitumor activity of the resultant compounds. Incorporation of electron-donating groups,such as methyl (as in 3f and 3g ) and methoxy (as in 3i- 3k) groups,in the benzene ring led to a decrease of the antitumor activity against both cell lines. Further studies will focus on structural optimization and structure-activity relationships of this class of compounds.
| Table 2 The in vitro antitumor activity against MCF-7 and HepG2 for title compounds 3a- 3k. |
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
A series of novel 2-trifluoromethylthieno[2, 3, d]pyrimidine derivatives were synthesized by a facile three-step procedure. The procedure exhibits several advantages,such as mild reaction conditions,simple protocol,and good yields. Their structures were characterized by IR,1H NMR,19F NMR,EI-MS and elemental analysis,and the structure of 3h was further elucidated by singlecrystal X-ray diffraction. The preliminary bioassay results imply that some of the compounds exhibit excellent antitumor activity against MCF-7 and HepG2 cells. These compounds will be further studied in future research. Acknowledgment
This work was financially supported by the National Natural Science Foundation of China (No. 21262012) and the Open Fund of Key Laboratory of Biologic Resources Protection and Utilization of Hubei Province (No. PKLHB1314).
| [1] | R. Adepu, D. Rambabu, B. Prasad, et al., Novel thieno[2,3-d]pyrimidines: their design, synthesis, crystal structure analysis and pharmacological evaluation, Org. Biomol. Chem. 10 (2012) 5554-5569. |
| [2] | J.G. Paez, P.A. Janne, J.C. Lee, et al., EGFR mutations in lung cancer: correlation with clinical response to gefitinib therapy, Science 304 (2004) 1497-1500. |
| [3] | B. Higgins, K. Kolinsky, M. Smith, et al., Antitumor activity of erlotinib (OSI-774, Tarceva) alone or in combination in human non-small cell lung cancer tumor xenograft models, Anticancer Drugs 15 (2004) 503-512. |
| [4] | Z.A. Liu, A. Fusi, A. Schmittel, et al., Eradication of EGFR-positive circulating tumor cells and objective tumor response with lapatinib and capecitabine, Cancer Biol. Ther. 10 (2010) 860-864. |
| [5] | Y. Dai, Y. Guo, R.R. Frey, et al., Thienopyrimidine ureas as novel and potent multitargeted receptor tyrosine kinase inhibitors, J. Med. Chem. 48 (2005) 6066-6083. |
| [6] | J.C. Aponte, A.J. Vaisberg, D. Castillo, et al., Trypanoside, anti-tuberculosis, leishmanicidal, and cytotoxic activities of tetrahydrobenzothienopyrimidines, Bioorg. Med. Chem. 18 (2010) 2880-2886. |
| [7] | N.S. Habib, R. Soliman, A.A. El-Tombary, S.A. El-Hawash, O.G. Shaaban, Synthesis and biological evaluation of novel series of thieno[2,3-d]pyrimidine derivatives as anticancer and antimicrobial agents, Med. Chem. Res. 22 (2013) 3289-3308. |
| [8] | B.Q. Cai, H.X. Jin, X.J. Yan, P. Zhu, G.X. Hu, 3D-QSAR and 3D-QSSR studies of thieno[2,3-d]pyrimidin-4-yl hydrazone analogues as CDK4 inhibitors by CoMFA analysis, Acta Pharmacol. Sin. 35 (2014) 151-160. |
| [9] | W.K. Hagmann, The many roles for fluorine in medicinal chemistry, J. Med. Chem. 51 (2008) 4359-4369. |
| [10] | X.J. Song, Z.C. Duan, Y. Shao, X.G. Dong, Facile synthesis of novel fluorinated thieno[2,3-d]pyrimidine derivatives containing 1,3,4-thiadiazole, Chin. Chem. Lett. 23 (2012) 549-552. |
| [11] | T. Horiuchi, M. Nagata, M. Kitagawa, K. Akahane, K. Uoto, Discovery of novel thieno[2,3-d]pyrimidin-4-yl hydrazone-based inhibitors of Cyclin D1-CDK4: synthesis, biological evaluation and structure-activity relationships. Part 2, Bioorg. Med. Chem. Lett. 17 (2009) 7850-7860. |
| [12] | A.G. Golub, V.G. Bdzohla, N.V. Briukhovetska, et al., Synthesis and biological evaluation of substituted (thieno[2,3-d]pyrimidin-4-ylthio)carboxylic acids as inhibitors of human protein kinase CK2, Eur. J. Med. Chem. 46 (2011) 870-876. |
| [13] | T. Beckers, A. Sellmer, E. Eichhorn, et al., Novel inhibitors of epidermal growth factor receptor: (4-(arylamino)-7H-pyrrolo[2,3-d]pyrimidin-6-yl)(1H-indol-2-yl)methanones and (1H-indol-2-yl)(4-(phenylamino)thieno[2,3-d]pyrimidin-6-yl)methanones, Bioorg. Med. Chem. 20 (2012) 125-136. |
| [14] | S.E. Abbas, N.M.A. Gawad, R.F. George, Y.A. Akar, Synthesis, antitumor and antibacterial activities of some novel tetrahydrobenzo[4,5]thieno[2,3-d]pyrimidine derivatives, Eur. J. Med. Chem. 65 (2013) 195-204. |
| [15] | P. Yang, Y. Wang, Z.C. Duan, et al., Synthesis, crystal structure, and biological activity of 4-chlorobenzaldehyde (2-trifluoromethyl-5,6,7,8-tetrahydrobenzo[ 4,5]thieno[2,3-d]pyrimidin-4-yl)hydrazone monohydrate, Chin. J. Struct. Chem. 32 (2013) 1023-1030. |
| [16] | T. Horiuchi, J. Chiba, K. Uoto, T. Soga, Discovery of novel thieno[2,3-d]pyrimidin-4-yl hydrazone-based inhibitors of Cyclin D1-CDK4: synthesis, biological evaluation, and structure-activity relationships, Bioorg. Med. Chem. Lett. 19 (2009) 305-308. |
| [17] | R.J. Gillespie, D.R. Adams, D. Bebbington, et al., Antagonists of the human adenosine A2A receptor. Part 1: discovery and synthesis of thieno[3,2-d]pyrimidine-4-methanone derivatives, Bioorg. Med. Chem. Lett. 18 (2008) 2916-2919. |
| [18] | M. Hayakawa, H. Kaizawa, H. Moritomo, et al., Synthesis and biological evaluation of 4-morpholino-2-phenylquinazolines and related derivatives as novel PI3 kinase p110a inhibitors, Bioorg. Med. Chem. 14 (2006) 6847-6858. |
| [19] | M.R. Prasad, D.P. Kishore, Multistep, microwave assisted, solvent free synthesis and antibacterial activity of 6-substituted-2,3,4-trihydropyrimido[1,2-c]9,10,11,12-tetrahydrobenzo[b]thieno[3,2-e]pyrimidines, Chem. Pharm. Bull. 55 (2007) 776-779. |
| [20] | A.E. Kassab, E.M. Gedawy, Synthesis and anticancer activity of novel 2-pyridyl hexahyrocyclooctathieno[2,3-d]pyrimidine derivatives, Eur. J. Med. Chem. 63 (2013) 224-230. |
| [21] | M. Sridhar, R.M. Rao, N.H.K. Baba, R.M. Kumbhare, Microwave-accelerated Gewald reaction: synthesis of 2-aminothiophenes, Tetrahedron Lett. 48 (2007) 3171-3172. |
| [22] | R. Rajagopal, T.M. Jyothi, T. Daniel, K.V. Srinivasan, B.S. Rao, Calcined Mg-Al hydrotalcite as a heterogeneous base catalyst for Gewald aminothiophene synthesis, Synth. Commun. 31 (2001) 3113-3117. |
| [23] | T. Wang, X.G. Huang, J. Liu, et al., An efficient one-pot synthesis of substituted 2-aminothiophenes via three-component Gewald reaction catalyzed by L-proline, Synlett (2010) 1351-1354. |
| [24] | D.D. Zhao, L. Li, F. Xu, Q. Wu, X.F. Lin, Bovine serum albumin-catalyzed one-pot synthesis of 2-aminothiophenes via Gewald reaction, J. Mol. Catal. B-Enzym. 95 (2013) 29-35. |
| [25] | R. Tayebee, F. Javadi, G. Argi, Easy single-step preparation of ZnO nano-particles by sedimentation method and studying their catalytic performance in the synthesis of 2-aminothiophenes via Gewald reaction, J. Mol. Catal. A-Chem. 368-369 (2013) 16-23. |
| [26] | E. Rezaei-Seresht, R. Tayebee, M. Yasemi, KG-60-piperazine as a new heterogeneous catalyst for Gewald three-component reaction, Synth. Commun. 43 (2013) 1859-1864. |
| [27] | P. Malherbe, R. Masciadri, R.D. Norcross, H. Ratni, A.W. Thomas, Thieno-pyridine derivatives as allosteric enhancers of the GABAB receptors, US2006135552 (2006). |
| [28] | G.M. Sheldrick, A short history of SHELX, Acta Cryst. A64 (2008) 112-122. |
| [29] | M.C. Alley, D.A. Scudiero, A. Monks, et al., Feasibility of drug screening with panels of human tumor cell lines using a microculture tetrazolium assay, Cancer Res. 48 (1988) 589-601. |