




b Department of Radiation Oncology, Ji'nan Military General Hospital, Jinan 250031, China
The use of effective antitumor agents derived from natural products is a promising strategy for cancer chemotherapy [1]. It is reported that more than 50% of antitumor drugs have been developed from natural products or their synthetic derivatives [2, 3]. Antitumor natural products also provide novel probes for chemical biology and target identification studies. For example, camptothecin (target: DNA topoisomerase I,Top1) and taxol (target: microtubule) represent two historic achievements in antitumor drug discovery [4].
Evodiamine is a quinazolinocarboline alkaloid with diverse biological activities which was isolated from the fruits of the traditional Chinese herbEvodiae fructus(Chinese name: Wu-ChuYu) [5]. In particular,evodiamine showed anti-proliferative activities against a wide variety of cancer cells by inducing apoptosis [6]. It is a good antitumor lead compound because of its broad-antitumor spectrum,low toxicity,and suitable physicochemical properties. In our previous studies,evodiamine was identified as a Top1 inhibitor by structure-based virtual screening [7]. Further structural optimization studies identified several highly active evodiamine derivatives which showed potentin vitro andin vivo antitumor activities [8].In silico target identification and biological assays revealed that the evodiamine derivatives were dual inhibitors of Top1 and topoisomerase II (Top2) [8].
Inspired by these encouraging results,herein a series of E-ring modified evodiamine derivatives were designed and synthesized. Compound 12 showed good antitumor activity with a broad spectrum. Its binding modes with topoisomerase I and II were clarified by molecular docking. 2. Experimental
Nuclear magnetic resonance (NMR) spectra were recorded on Bruker AVANCE300 and AVANCE500 spectrometers (Bruker Company,Germany),with TMS as an internal standard and DMSO-d6as the solvent. Chemical shifts (δ values) and coupling constants (∫ values) are expressed in ppm and Hz,respectively. ESI mass spectra were performed on an API-3000 LC-MS spectrometer. TLC analysis was carried out on silica gel plates GF254 (Qindao Haiyang Chemical,China). Silica gel column chromatography was performed with Silica gel 60G (Qindao Haiyang Chemical,China). Commercial solvents were used without any pretreatment. 2.1. Chemistry
The synthetic route of E-ring modified evodiamine derivatives is described in Scheme 1. 3,4-Dihydro- &bate;-carboline (4) was prepared by reacting tryptamine 2 with ethyl formate,followed by intramolecular ring closure in the presence of POCl3. Substituted 2-aminobenzoic acid (5) was treated with triphosgene to provide isatoic anhydride,which was methylated by CH3I to afford compound 7 [8]. Condensation of 3,4-dihydro- &bate;-carboline (4)with N-methyl isatoic anhydride 7 provided E-ring derivatives 8a-c. Reduction of the nitro group of compounds 8a and 8dusing 10% Pd/C and H2 afforded the amino derivatives 9aand 9b. Compound 9awas further acylated to give compound 10,which was subsequently treated with Na2S to provide compound 11. The Suzuki-[Miyaura reaction of compound 8c with 2-(1-piperazinyl)pyridine-4-boronic acid pinacol ester afforded the target compound 12.

|
Download:
|
| Scheme 1. Synthetic routes of the target compounds. Reagents and conditions: (a) Ethyl formate, reflux, 6 h, yield 100%; (b) POCl3,CH2Cl2,0°C, 6 h, yield 85%; (c) Triphosgene, THF, reflux, 4 h, yield 90%-95%; (d) CH3I, KOH,DMF, rt, 2 h, yield 60–87%; (e) CH2Cl2,45°C, 6 h, yield 50%-62%; (f) 10% Pd/C, H2 ,DMF, r.t., 12 h, yield 11–73%; (g) 2-bromoacetyl bromide, Et3N, CH2Cl2,0°C, 1 h, yield 84%; (h) Na2S·9H2 O,DMF, r.t., 12 h, yield 70%; (i) 2-(1-piperazinyl)pyridine-4-boronic acid pinacol ester, Pd(pph3)4,Na2CO3,H2 O, reflux, 3.5 h, yield 51%. | |
Synthesis of 3-nitroevodiamine ( 8a ): A solution of 3,4-dihydro &bate;-carboline [8] (4,0.17 g,1 mmol) and 1-methyl-6-nitro-1H-benzo[d][1, 3]oxazine-2,4-dione [8] (7a,0.2 g,1 mmol) in CH2Cl2 (15 mL) were heated to 45°C and stirred for 6 h. Then the reaction mixture was concentrated to one-half of its original volume. The precipitate was collected by filtration and washed with alcohol to yield the product 8a (0.21 g,62%) as yellow solid. 1H NMR (300 Hz,DMSO-d6):δ2.68-2.70 (m,1 H),2.95-3.01 (m,2H),3.45 (s,3H),4.60-4.64 (m,1H),6.50 (s,1H),6.96-7.11 (m,4H),7.32 (d, 1H,J= 7.8 Hz),7.42 (d,1H,J= 7.8 Hz),8.24 (dd,1H,J= 9.0 Hz, 2.7 Hz),8.43 (d,1H,J= 2.7 Hz),10.89 (s,1H). ESI-MS: Calcd. for C19H16N4O3[M-H]-:m/z347.12; found: 347.76.
The synthetic method for target compounds 8d-c was similar to that of compound 8a .
2-Chloro-3-nitroevodiamine ( 8d): Yellow solid (0.33 g,yield: 50%). 1H NMR (300 Hz,DMSO-d6):δ2.68-2.73 (m,1H),2.95-3.01 (m,2H),3.46 (s,3H),4.61-4.67 (m,1H),6.52 (s,1H),7.00-7.16 (m,3H),7.35 (d,1H,J= 7.5 Hz),7.43 (d,1H,J= 7.5 Hz),8.37 (s,1H), 10.90 (s,1H). ESI-MS: Calcd. for C19H14ClN4O3 [M-H]-: m/z 381.79; found: 381.81.
3-Iodoevodiamine (8c): White solid (0.07 g, yield: 50%). 1H NMR (500 MHz, DMSO-d6): δ 2.75–2.78 (m, 1H), 2.91–2.92 (m, 1H), 2.96 (s, 3H), 3.19–3.21 (m, 1H), 4.58–4.62 (m, 1H), 6.17 (s, 1H), 6.87 (d, 1H,J= 8.6 Hz), 7.01 (t, 1H,J= 7.6 Hz), 7.11 (t, 1H, J= 7.6 Hz), 7.35 (d, 1H,J= 8.1 Hz), 7.46 (d, 1H,J= 8.1 Hz), 7.45 (dd, 1H, J= 8.6 Hz, 2.1 Hz), 7.98 (d, 1H,J= 2.1 Hz), 11.03 (s, 1H). ESI-MS: Calcd. for C19H17IN3O[M+H]+ :m/z430.26; found: 430.39.
Synthesis of 3-aminoevodiamine ( 9a): To a stirring solution of 8a (0.12 g,0.34 mmol) inDMF (10 mL),10% Pd/C (0.01 g) was added. The reaction mixture was stirred under hydrogen atmosphere for 12 h at room temperature. Then the mixture was diluted with water (50 mL) and extracted with EtOAc (3×50 mL). The combined organic layers were washed with saturated sodium chloride solution (3×50 mL),dried over anhydrous Na2SO4 and concentrated under reduced pressure. The residue was purified by silica gel column chromatography (CH2Cl2-MeOH,gradient 100:1, 100:2) to give compound 9a(0.08 g,73%) as a yellow solid. 1H NMR (500 Hz,DMSO-d6):δ 2.27 (s,3H),2.77-2.79 (m,1H),2.88-2.92 (m,1H),3.11-3.13 (m,1H),4.63-4.67 (m,1H),5.10 (s,2H),5.90 (s,1H),6.78 (dd,1H,J= 8.5 Hz,2.5 Hz),6.94 (d,1H,J= 8.5 Hz),7.01 (t,1H,J= 7.5 Hz),7.10-7.14 (m,2H),7.36 (d,1H,J= 8.0 Hz),7.51 (d,1H,J= 8.0 Hz),11.26 (s,1H). ESI-MS: Calcd. for C19H19N4O [M+H]+:m/z319.15; found: 319.93.
The synthetic method for target compounds 9bwas similar to that of compound 9a.
2-Chloro-3-aminoevodiamine ( 9b): Red solid (0.09 g,yield: 11%). 1H NMR (500 Hz,DMSO-d6):δ 2.27 (s,3H),2.73-2.77 (m, 2H),3.11-3.13 (m,1H),4.63-4.66 (m,1H),5.10 (s,2H),5.90 (s,1H),6.78 (dd,1H,J= 8.5 Hz,2.5 Hz),6.94 (d,1H,J= 8.5 Hz),7.01 (t,1H, J= 7.5 Hz),7.10-7.14 (m,2H),7.36 (d,1H,J= 8.0 Hz),11.26 (s,1H). ESI-MS: Calcd. for C19H18ClN4O [M+H]+: m/z 353.83; found: 353.49.
Synthesis of 2-bromoacetamidoevodiamine (10): To a stirring solution of 9a(0.1 g,0.3 mmol) and Et3N (0.04 mL,0.3 mmol) in CH2Cl2(20 mL),2-bromoacetyl bromide (0.04 mL,0.47 mmol) was added and stirred for 1 h at 0°C. The solvent was removed under reduced pressure. The residue was purified by silica gel column chromatography (CH2Cl2:MeOH = 100:1,v/v) to afford10(0.11 g, yield 84%) as yellow solid. 1H NMR (300 Hz,DMSO-d6):δ2.69 (s, 3H),2.85 (m,2H),3.15 (m,1H),4.01 (s,2H),4.62 (m,1H),6.07 (s, 1H),7.00 (t,1H,J= 7.8 Hz),7.08-7.13 (m,2H),7.35 (d,1H, J= 7.8 Hz),7.48 (d,1H,J= 7.8 Hz),7.73 (dd,1H,J= 9.0 Hz,2.4 Hz), 8.03 (d,1H,J= 2.7 Hz),10.41 (s,1H),11.16 (s,1H). ESI-MS: Calcd. for C21H19BrN4O2[M+H]+:m/z439.07; found: 439.75. Synthesis of 2-mercaptoacetamidoevodiamine (11): A solution of10(0.05g,0.11mmol)and Na2S·9H2 O (0.27 g,1.1 mmol) in DMF (5 mL) was stirred at room temperature for 12 h. The solvent was removedin vacuoand the residue was diluted with water and filtered from insolubles. The filtrate was acidified topH 1.2 with 3 mol/L HCl. The precipitate was collected and purified by silica gel column chromatography (CH2Cl2-MeOH,gradient 100:1- 100:3) to give compound11(0.03 g,yield 70%) as a yellow solid. 1H NMR(300Hz,DMSO-d6): d1.30 (s,1 H),2.66 (s,3H),2.86 (m,2H),3.20(m,1H),3.48(s,2H),4.64(m,1H),6.06(s,1H),7.01 (t,1H,J= 7.2 Hz),7.10-7.15 (m,2H),7.36 (d,1H,J=8.1Hz),7.50 (d,1H,J= 8.1 Hz),7.73 (d,1H,J= 9.3 Hz),8.07 (s,1H),10.19 (s,1H), 11.19 (s,1H). ESI-MS: Calcd. for C21H20N4O2S[M+H]+: m/z 393.14; found: 393.84.
Synthesis of 3-(2-(piperazin-1-yl)pyridin-4-yl)evodiamine (12): Under an nitrogen atomosphere, 8c (0.1 g,0.23 mmol),2-(1-piperazinyl)pyridine-4-boronic acid pinacol ester (0.1 g, 0.35 mmol) and tetrakistriphenylphosphine palladium (40 mg) were dissolved inDMF (5 mL). An aqueous sodium carbonate solution (2 mol/L,1 mL) was added and the mixture was heated under reflux for 3.5 h. After cooled to room temperature,the reaction mixture was diluted with water (50 mL) and then extracted with EtOAc (3×50 mL). The combined organic layers were washed with saturated sodium chloride solution (3×50 mL), dried over anhydrous Na2SO4 and concentrated under reduced pressure. The residue was purified by silica gel column chromatography (CH2Cl2:MeOH = 100:1,v/v) to give compound12(0.056 g, yield 51%) as a yellow solid. 1H NMR (300 Hz,DMSO-d6):d2.15 (s,3H),2.82 (m,1H),3.01 (m,1H),3.16 (m,1H),3.74 (s,3H),3.81 (s,3H),4.64 (m,1H),6.25 (s,1H),6.85 (d,1H,J= 9.3 Hz),6.92 (s,1H), 6.99 (t,1H,J= 7.8 Hz),7.05-7.11 (m,2H),7.34 (d,1H,J= 7.8 Hz), 7.44 (d,1H,J= 7.8 Hz),7.85 (dd,1H,J= 8.7 Hz,2.1 Hz),8.04 (d,1H, J= 2.1 Hz),8.09 (d,1H,J= 5.1 Hz),10.99 (s,1H). ESI-MS: Calcd. for C28H28N6O[M+H]+:m/z465.24; found: 465.84. 2.2. Biological assay
Top1-mediated supercoiled pBR322 relaxation assay: Relaxation assays were carried out as previously described [7, 8]. The reaction buffer contained 35 mmol/L Tris-HCl (pH 8.0),72 mmol/L KCl,5 mmol/L MgCl2,5 mmol/L dithiothreitol,5 mmol/L spermidine,0.1% bovine serum albumin (BSA),0.25 ug pBR322 plasmid DNA,and 1 unit of Top1 (TaKaRa Biotechnology Co.,Ltd.,Dalian) in a total volume of 20 uL. After incubation at 37°C for 15 min,the reaction was stopped by the addition of 2 uL of 10× loading buffer (0.9% sodium dodecyl sulfate (SDS),0.05% bromophenol blue,and 50% glycerol). Then,the DNA samples were subjected to electrophoresis in 0.8% agarose gel in TAE (Tris-acetate-EDTA) at 8 V/cm for 1 h. Gels were stained with ethidium bromide (0.5mg/mL) for 60 min. The DNA band was visualized over UV light and photographed with Gel Doc Ez imager software (Bio-Rad Laboratories Ltd.).
Top2 α-mediated supercoiled pBR322 relaxation assay: Relaxation assays were performed using Topoisomerase II Drug Screening Kits (TopoGEN,Inc.) [8]. The reaction buffer contained 50 mmol/L Tris-HCl (pH 8.0),150 mmol/L NaCl,10 mmol/L MgCl2,5 mmol/L dithiothreitol,30 ug/mL bovine serum albumin (BSA),2 mmol/L ATP,0.25mg pBR322 plasmid DNA,and 0.75 unit of Top2 α (TopoGEN,Inc.) in a total volume of 20mL. After incubation at 37°C for 30 min,the reaction was terminated by the addition of 2mL 10×gel loading buffer (0.25% bromophenol blue,50% glycerol). The reaction products were analyzed on 1% agarose gel at 8 V/cm for 1 h with TAE (Tris-acetate-EDTA) as the running buffer. Gels were stained with ethidium bromide (0.5mg/mL) for 60 min. The DNA band was visualized over UV light and photographed with Gel Doc Ez imager software (Bio-Rad Laboratories Ltd.).
In vitro cytotoxicity assay: Cells were seeded in 96-well microtiter plates at a density of 5×103/well and treated in triplicate with gradient concentrations of compounds in a humidified atmosphere with 5% CO2 at 37°C for 72 h. 20mLof MTT (3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide) solution (5 mg/mL) was added to each well and the plate was incubated for an additional 4 h. The formazan was dissolved in 100 uL of DMSO. The absorbance (OD) was read on a Wellscan MK-2 microplate reader (Labsystems) at 570 nm. The concentration causing 50% inhibition of cell growth (IC50) was determined by the Logit method [9, 10]. All experiments were performed three times. 2.3. [M+H]+olecular docking
In silicodocking was performed using GOLD 5.1 [11] on a Linux PC as described before [8]. The X-ray crystallographic structures of CPT-DNA-Top1 ternary complex (PDB code: 1T8I [12]) and ATPase domain of human Top2 α(PDB code: 1ZXM[13]) were obtained from the Protein Data Bank. They were prepared for molecular docking analysis in Discovery Studio 3.0 [14] by removing the ligand fromthe binding pocket,merging nonpolar hydrogens,adding polar hydrogens,and rendering Gasteiger charges to each atom. 3. Results and discussion
3.1. In vitro antitumor activity
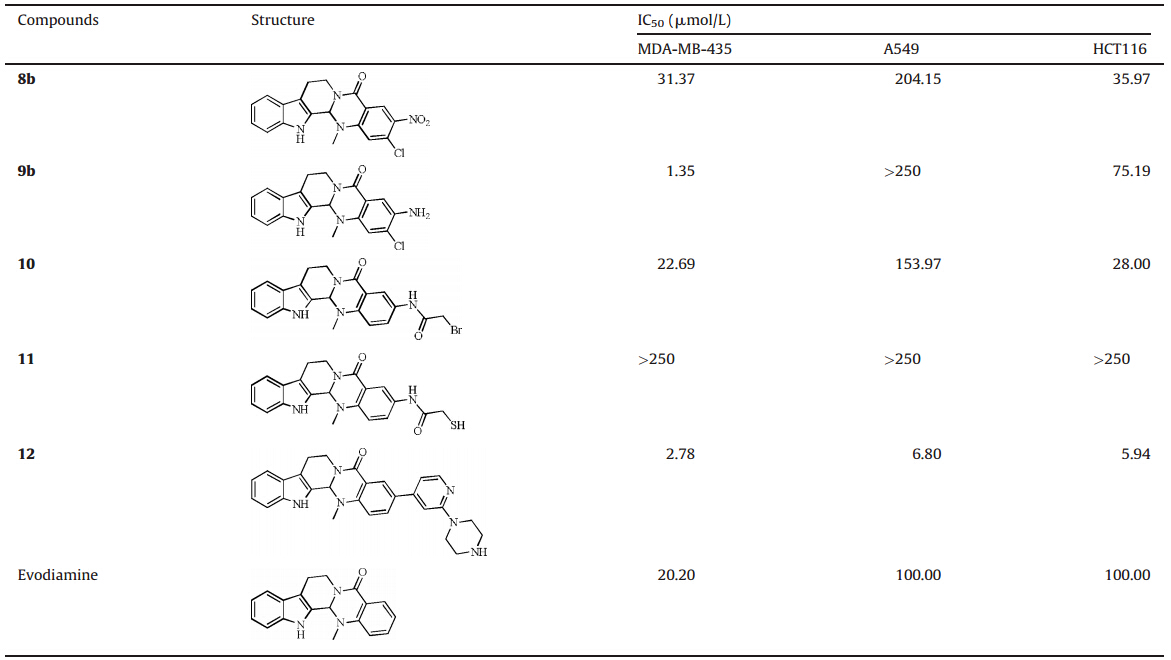
The growth inhibitory activities of the target compounds toward human cancer cell lines A549 (lung cancer),MDA-MB-435 (breast cancer),and HCT116 (colon cancer) were determined using MTT assay. Evodiamine was used as the reference drug. When the 2-bromoacetamide group was attached on the position 3 of evodiamine E-ring,compound 10 showed moderate in vitro antitumor activity against MDA-MB-435 (IC50=22.69 umol/L), A549 (IC50=153.97 umol/L) and HCT 116 (IC50=28 umol/L) cell lines (Table 1). However,the loss of the antitumor activity was observed for the corresponding thio-derivative11. 2-Chloro-3-nitro evodiamine ( 8d) also showed moderate antitumor activity against all three of the tested cancer cell lines. When its nitro group was reduced to the amino group,compound 9bshowed significantly improved activity against the MDA-MB-435 cell line (IC50=1.35 umol/L). Interestingly,the compound with a 2-(piperazin-1-yl)pyridyl group at the position 3 of evodiamine E-ring (compound 12) showed good antitumor activity with a broad spectrum. The IC50values of compound 12a gainst MDA-MB-435, A549 and HCT 116 cell lines were 2.78 umol/L,6.80mmol/L and 5.94mmol/L,respectively. As compared with evodiamine,most of the target compounds showed better antitumor activity and broader spectra.
| Table 1 In vitro antitumor activity of evodiamine derivatives (IC50,mmol/L). |
{kind=link}
[M+H]+uman Top2 can be classified into two subgroups,Top2α and Top2&bate;.Top2α increases its expression during the cell cycle and mitosis,and its concentration is much higher in tumor cells. Top2&bate; expression remains relatively constant and at low level throughout the cell cycle [15, 16]. Thus,Top2α is a good target for antitumor drug discovery. Previously,evodiamine and its derivatives were identified as dual Top1/Top2 inhibitors [8]. Herein,Top1- and Top2α mediated pBR322 DNA relaxation assays with purified Top1 and Top2α were used to investigate the inhibitory activity of compound 12 with its concentrations from 10umol/L to 100umol/L. Camptothecin (CPT) and etoposide (Eto) were employed as positive controls. As shown in Fig. 1A,compound 12 inhibited Top1-mediated relaxation of pBR322 DNA in a concentration-dependent manner. It showed moderate inhibitory activity at 100 umol/L and lower activity at 50 umol/L. The inhibitory activity of compound 12 was much higher than evodiamine,which only showed marginal activity at 100 umol/L [7]. Compound12was also proven to be a potent inhibitor of the catalytic activity of Top2α (Fig. 1B). Particularly,it showed comparable inhibitory activity against Top2-mediated DNA relaxation to that of Eto at 100mmol/L and 50mmol/L,respectively.

|
Download:
|
| Fig. 1. Top1 and Top2 inhibitory activity of compound 12. (A) Inhibition of Top1 relaxation activity ranging from 10mmol/L to 100µmol/L. Lane 1,supercoiled plasmid DNA; Lane 2,DNA + Top1; Lanes 3-5,DNA + Top1 + CPT (100,50 and 10mmol/L); Lanes 6-8,DNA + Top1 + 12(100,50 and 10mmol/L). (B) Inhibition of Top2 relaxation activity ranging from 10mmol/L to 100mmol/L. Lane 1,supercoiled plasmid DNA; Lane 2,DNA + Top2; Lanes 3-5,DNA + Top1 + Eto (100,50 and 10mmol/L); Lanes 6-8,DNA + Top2+ 12 (100,50 and 10mmol/L). Lanes order from left to right. | |
{kind=link}
In an attempt to understand the binding mode of compound 12 to Top1-DNA complex and ATP-binding domain of Top2α ,docking simulation was performed. As indicated in the interaction model between 12 and Top1-DNA complex (Fig. 2A),its E-ring and pyridyl group intercalates at the site of DNA cleavage and formsp-stacking interactions with both the -1 (upstream) and +1 (downstream) base pairs. The N atom of pyridine forms a hydrogen bond with the hydroxyl group of Thr718. AB-ring is faced into the major groove and its indole N[M+H]+ undergoes a strong hydrogen bond interaction with the oxygen atom of Guanine. Moreover,compound 12 is observed to be well-fitted into the ATP-binding domain of Top2α (Fig. 2B). It is capable of making four hydrogen-bonds to enhance the stabilization of the 12-ATPase complex. The N atom of pyridine is shown to participate in hydrogen bonding interactions with Ala167 and Lys168,respectively. The amino group on the piperazine also can form two hydrogen bonds with backbone amide of Arg162 and Asn163. ChemPLP score was also calculated to evaluate the binding affinity. The most active compound 12 showed the highest binding affinity to Top1 and Top2α with ChemPLP sore 82.54 and 61.00,respectively. In contrast,removal of the piperazine group led to the decrease of the binding affinity (ChemPLP sore: 68.04 for Top1 and 54.48 for Top2α ). Evodiamine showed the lowest ChemPLP scores (Top1: 64.45,Top2α : 53.34). The computational results were consistent with the biological activities and highlighted the importance of the pyridine and piperazine group for Top1 and Top2α binding.

|
Download:
|
| Fig. 2. Binding mode with Top1/Top2. (A) Binding mode of 12 to Top1-DNA complex. (B) Binding mode of 12 to ATP-binding domain of Top2. The figure was generated using PyMol (http://www.pymol.org/). | |
{kind=link}
In summary,a series of novel E-ring modified evodiamine derivatives were designed and synthesized as antitumor agents. Top1/Top2 inhibitory assay and further molecular docking confirmed that the derivatives acted by dual inhibition of Top1 and Top2. As compared with evodiamine,most of the compounds showed better antitumor activity and broader spectra. Compound 12showed good antitumor activity with an IC50 ranging from 2.78 umol/L to 5.94mmol/L. It can be used as a good starting point for further optimization. Acknowledgments
This work was supported by National Natural Science Foundation of China (Nos. 81222044,81373278),Key Project of Science and Technology of Shanghai (No. 11431920402),the National Basic Research Program of China (No. 2014CB541800),Shanghai RisingStar Program (No. 12QH1402600),and Shanghai Municipal Health Bureau (No. XYQ2011038).
| [1] | G.M. Cragg, P.G. Grothaus, D.J. Newman, Impact of natural products on developing new anti-cancer agents, Chem. Rev. 109 (2009) 3012-3043. |
| [2] | D.J. Newman, G.M. Cragg, Natural products as sources of new drugs over the 30 years from 1981 to 2010, J. Nat. Prod. 75 (2012) 311-335. |
| [3] | D.J. Newman, G.M. Cragg, Natural products as sources of new drugs over the last 25 years, J. Nat. Prod. 70 (2007) 461-477. |
| [4] | N.H. Oberlies, D.J. Kroll, Camptothecin and taxol: historic achievements in natural products research, J. Nat. Prod. 67 (2004) 129-135. |
| [5] | J. Jiang, C. Hu, Evodiamine: a novel anti-cancer alkaloid from Evodia rutaecarpa, Molecules 14 (2009) 1852-1859. |
| [6] | Z.G. Yang, A.Q. Chen, B. Liu, Antiproliferation and apoptosis induced by evodiamine in human colorectal carcinoma cells (COLO-205), Chem. Biodivers. 6 (2009) 924-933. |
| [7] | G. Dong, C. Sheng, S. Wang, et al., Selection of evodiamine as a novel topoisomerase I inhibitor by structure-based virtual screening and hit optimization of evodiamine derivatives as antitumor agents, J. Med. Chem. 53 (2010) 7521-7531. |
| [8] | G. Dong, S. Wang, Z. Miao, et al., New tricks for an old natural product: discovery of highly potent evodiamine derivatives as novel antitumor agents by systemic structure-activity relationship analysis and biological evaluations, J. Med. Chem. 55 (2012) 7593-7613. |
| [9] | H. Huang, Q. Chen, X. Ku, et al., A series of alpha-heterocyclic carboxaldehyde thiosemicarbazones inhibit topoisomerase IIa catalytic activity, J. Med. Chem. 53 (2010) 3048-3064. |
| [10] | L. Du, H.C. Liu, W. Fu, et al., Unprecedented citrinin trimer tricitinol B functions as a novel topoisomerase IIa inhibitor, J. Med. Chem. 54 (2011) 5796-5810. |
| [11] | G. Jones, P. Willett, R.C. Glen, et al., Development and validation of a genetic algorithm for flexible docking, J. Mol. Biol. 267 (1997) 727-748. |
| [12] | B.L. Staker, M.D. Feese, M. Cushman, et al., Structures of three classes of anticancer agents bound to the human topoisomerase I-DNA covalent complex, J. Med. Chem. 48 (2005) 2336-2345. |
| [13] | H. Wei, A.J. Ruthenburg, S.K. Bechis, G.L. Verdine, Nucleotide-dependent domain movement in the ATPase domain of a human type IIA DNA topoisomerase, J. Biol. Chem. 280 (2005) 37041-37047. |
| [14] | Accelrys Software Inc., Discovery Studio Modeling Environment, Release 3.0, Accelrys Software Inc., San Diego, 2010. |
| [15] | K. Chikamori, A.G. Grozav, T. Kozuki, et al., DNA topoisomerase II enzymes as molecular targets for cancer chemotherapy, Curr. Cancer Drug. Targets 10 (2010) 758-771. |
| [16] | A.T. Baviskar, C. Madaan, R. Preet, et al., N-fused imidazoles as novel anticancer agents that inhibit catalytic activity of topoisomerase IIa and induce apoptosis in G1/S phase, J. Med. Chem. 54 (2011) 5013-5030. |