




bState Key Laboratory of Elemento-Organic Chemistry, Institute of Elemento-Organic Chemistry, Nankai University, Tianjin 300071, China;
cSchool of Pharmaceutical Sciences, Sun Yat-Sen University, Guangzhou 510006, China
Compounds could aggregate through intermolecular noncovalent bonding interactions such as hydrogen bonds,electrostatic interactions,hydrophobic interactions,π-π interactions and so on,which contribute to different biological or chemical properties [1]. In drug discovery,some small molecules could aggregate into submicrometer particles,nonspecifically inhibit different enzymes [2, 3],or promote enzyme unfolding [4]. The aggregate size of compounds was also associated with their oral bioavailability for some hydrophobic molecules [5]. Thus,not only the molecular structure but also their aggregation states could affect the bioactivity of compounds.
For pesticide,the final outcome depends on the spraying, distribution,transportation,interaction with target and other processes,rendering the interpretation of thein vivo efficacy difficult. Many pesticides showed distinct bioactivityin vivoandin vitro,which can be explained by hydrophilicity [6, 7] but might also be influenced by the molecular aggregation states. In order to explore the relationship between aggregation state and bioactivity, the quantitative aggregation-activity relationship (QAAR) method was proposed in our earlier work,in which dimer was used as the simplest aggregation state,due to the complexity and difficulty in modeling the multimolecular aggregation states. QAAR modeling was performed on two series of benzoylphenylureas insect growth regulators [8, 9]. Better regression of the QAAR models indicated that the bioactivity might strongly correlate with the molecular aggregation states. However,the benzoylphenylureas compounds used in our previous research are of poor water solubility and their dimer-aggregate models are generated based on intermolecular hydrogen-bonds. Thus,the following two questions are still remained to be clarified: (1) Whether molecular aggregation properties relate to the bioactivity of pesticides with good solubility,and (2) whether other non-covalent interactions such asπ-π interactions could be used in building the aggregate models in a QAAR study.
To answer the two questions above,QAAR studies were
performed on sulfonylurea herbicides,inhibitors of acetohydroxyacid synthase (AHAS),with high efficiency,low toxicity and good environment compatibility [10, 11]. Some series of
the highly soluble sulfonylurea compounds had been reported,
such as N-[2-(4-methyl)pyrimidinyl]-N'-2-methoxycarbonylbenzene sulfonylurea [12, 13, 14, 15, 16]. QSAR studies based on monomeric structures had also been performed to analyze the effects of
substituents on bioactivity [17, 18, 19, 20, 21, 22, 23, 24, 25, 26, 27]. Interestingly,a stable intermolecular π-π stacking dimeric structural motif was observed in the crystal structure ofN-[2-(4-methyl)pyrimidinyl]-N'-2-methoxycarbonyl-benzene sulfonylurea [14]. In this
study,π-π stacking dimer models of 24 sulfonylurea compounds
were constructed,followed by QAAR and QSAR investigations
based on dimer-aggregate and monomer descriptors,respectively. To a certain degree,the QAAR and QSAR models elucidated the possible roles of aggregation state on the bioactivity of highly
soluble pesticide systems.
2. Materials and methods
2.1. Biological data sets
All the 24 sulfonylurea compounds reported in our previous
study [15] were used to develop the QAAR/QSAR models,20 of which were selected randomly as training set and the rest 4 were
used as the test set. All the structures and herbicidal activity were
listed in Table 1.
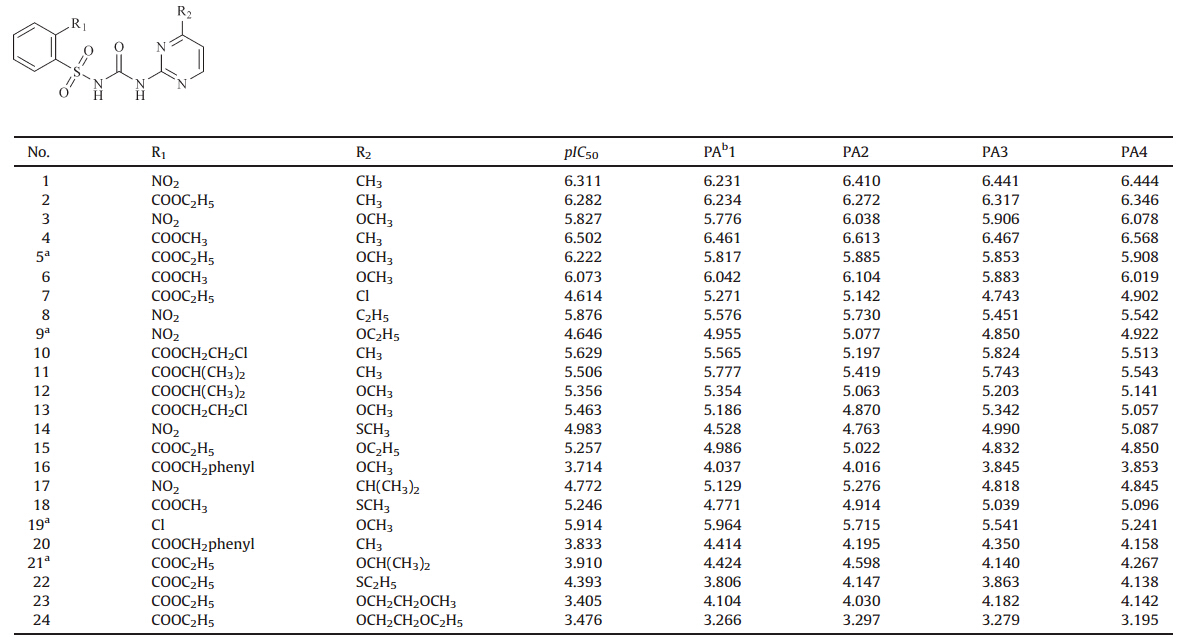
Based on the crystal structure ofN-[2-(4-methyl)pyrimidinyl]-N'-2-methoxycarbonyl-benzene sulfonylurea,the homo-dimeraggregate structures were established through π-π stacking
interactions (Fig. 1). The B97D/TZVP method,a density functional
theory (DFT) method that could produce more reliable results for
π-π interaction systems [28],was applied to optimize all monomeric and homo-dimeric structures. All calculations were
performed using the GAUSSIAN 09 software (Gaussian,Inc.,
Wallingford,CT,2009).
The optimized structures of monomeric and dimeric systems
were imported into databases of the Molecular Operating
Environment (MOE 2008.10). Then,more than 300 physicochemical descriptors were generated. The definitions of these descriptors included in the models are listed and described below. The
calculated descriptors were initially screened for invariant nature,
insignificance using QuaSAR Contingency module of MOE,which is
a statistical application designed to assist in the selection of
descriptors for QSAR. Further,the interrelation research was
performed to limit the number of descriptors for the study.
2.4. QAAR/QSAR modeling
Both monomer and aggregate based QSAR models were built
using the reduced descriptor pool as an independent variable and pIC50 as a dependent variable by forward stepwise regression
analyses. QSAR equations were acquired according to different
combinations of various descriptors. The data matrix was analyzed
using the partial least squares (PLS) method [29]. The quality of each regression model was evaluated,using a squared correlation
coefficient (r2),cross validation squared correlation coefficient (q2),
and root mean square error (RMSE). The correlation coefficientr2indicated how well the equation fits the data. The ‘leave-one-out’
(LOO) cross-validation coefficient q2was considered as an
indicator of the predictive performance and stability of a QSAR
model [30, 31].
3. Results and discussion
Theπ-π stacking aggregate models were generated by the
B97D/TZVP method. The optimized geometry of dimeric compound 4 was depicted in Fig. 1. The distance between the centroids
of two benzene rings was 3.75 Å ,which showed a good π-π
interaction. Then,QAAR/QSAR investigations were performed on
the descriptors calculated from the optimized aggregate/monomer
structures. The best established QSAR equations including two or
three monomeric descriptors were summarized in Eqs. (1) and (3)
respectively along with their statistical parameters,as well as their
corresponding QAAR Eqs. (2) and (4) based on aggregate
descriptors. The descriptors used in QSAR building were defined
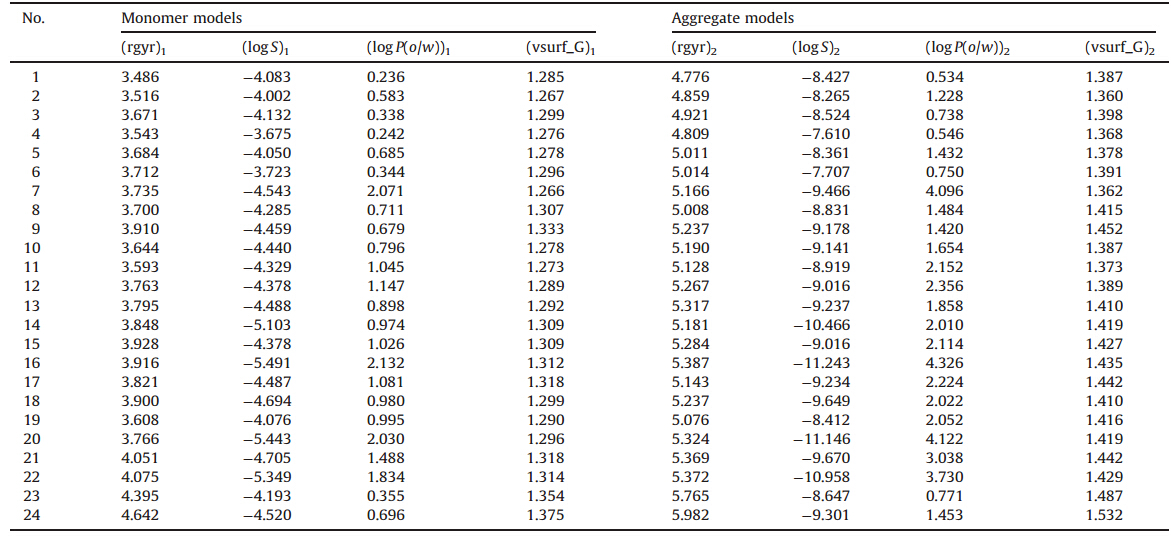
as follows: radius of gyration (rgyr),log of the aqueous solubility
(logS),log of the octanol/water partition coefficient (log P(o/w)),
surface globularity (vsurf_G).
Rgyr is a common QSAR descriptor,which is a measure of
molecular compactness for long chain molecules such as polymers.
It is calculated as the root mean square distance of the objects’
parts from either its center of gravity or a given axis. Small values
are obtained when most of the atoms are close to the center of
mass [32]. Rgyr is often used to describe the structural changes in the protein. The change of the Rgyr correlates to the degree that the
structure spreads out from its center [33]. Here,it was used to characterize the aggregation state changing from dimer to
monomer.
Log S is calculated from an atom contribution linear atom type
model [34]. The aqueous solubility of a compound significantly affects its absorption and distribution characteristics. Typically,
low solubility results in poor absorption. Aqueous solubility is
among the most important characteristics in ADME studies and the
relevant physicochemical descriptors in QSAR studies [32].
The log P(o/w) value is known as a measure of lipophilicity. In
drug research,partition coefficients are useful,for example,in
estimating distribution of drugs in the body [35]. LogPhas been
widely used to study biological processes relevant to drug action,
cellular uptake,metabolism,bio-availability,and toxicity [36]. Hydrophobic drugs with high octanol/water partition coefficients
are preferentially distributed to hydrophobic compartments such
as lipid bilayers of cells while hydrophilic drugs (low octanol/water
partition coefficients) are often found in hydrophilic compartments such as blood serum.
Vsurf_G is defined as the ratio of the molecular surface to the
surface area of a sphere of the same volume. Globularity is 1.0 for
perfect spherical molecules. It assumes values greater than 1.0 for
real spheroidal molecules [32]. It is specifically designed for the prediction of pharmacokinetic properties [37] and is also related to
molecular flexibility. In the QAAR/QSAR equations,nis the number of compounds in
the training set,r2is the correlation coefficient,q2is the LOO crossvalidated coefficient,RMSE is the root mean square error. A good
QAAR/QSAR model is indicated byr2andq2values close to 1.0,as
well as small RMSE. As a rule of thumb,the equations with
regression coefficients r2>0.80 and q2>0.50 are considered
reasonable. It can be found from Fig. 2 that all the equations
exhibited good regressions between the experimental and predicted activity.
The equations implied that the physical-chemical properties
logSand log P(o/w),which were independent of conformation,
rgyr and vsurf_G,which depended on the structure connectivity
and conformation,played important roles in bioactivity. All QAAR/
QSAR equations showed a negative correlation between rgyr and
activity. It indicated that smaller rgyr values may be associated
with higher activity. As shown in Table 2,the aggregate rgyr values
were smaller than the sum of those of two monomers,which
indicated the closer proximity between two monomers in the
aggregation state and more structural changes during the
deaggregation,which would increase the bioactivity. Similarly,
vsurf_G had a negative correlation with activity. Forming
aggregates reduced the values of vsurf_G,thereby,improved
activity. LogSand log P(o/w) showed opposite and negative sign in
the equations respectively,which meant that high solubility and
low octanol/water partition coefficients were favorable for activity.Taking together,the compounds that had high activity should have
small values of rgyr,vsurf_G,log P(o/w) and a large value of logS.
For example,compound 4 had the largest logSvalue,small rgyr
and vsurf_G values,and nearly the smallest log P(o/w) value,and
also had the highest activity. In contrast,compound 16 had the
lowest logSvalue,the biggest log P(o/w) value,large rgyr and
vsurf_G values,and compound 24 had the largest rgyr and vsurf_G
values,a small logSvalue,big log P(o/w) value. As a result,both
compounds 16 and 24 had weak activity.
All equations established here displayed a good predictive ability
and small errors between the predicted and experimental activity,
which indicated that the bioactivity of compounds deeply depended
on the properties of molecular aggregation state,as well as the
monomer. Furthermore,all aggregate-based QAAR equations
showed slightly better correlations and predictability than the
monomeric QSAR ones. Especially for the Eq. (4),which had three
aggregate descriptors,achieved the highestr2andq2values. It
revealed that,in herbicide system with high solubility,the
bioactivity was strongly correlated with the properties of molecular
aggregation state,which is consistent with the findings in our
previous study on poorly water-soluble insect growth regulators. In
the current study,π-π interactions instead of hydrogen bonds were
successfully used in the building of dimer-aggregate models,which
revealed thatπ-π interaction is also a satisfactory non-covalent
interaction that can be used in QAAR studies.
4. Conclusion
The QAAR/QSAR studies were carried out on a series of highly
water-soluble sulfonylurea herbicidal compounds using dimeric
and monomeric descriptors. Four QAAR and QSAR equations were
successfully established. The QAAR/QSAR models revealed that
low values of rgyr and vsurf_G for the formation and dissociation of
dimers,as well as high values of logSand low values of log P(o/w)
would lead to high bioactivity. Accordingly,the QAAR approach
was not only appropriate for poorly water-soluble insect growth
regulators,but also for highly water-soluble sulfonylurea herbicide,and not only hydrogen bonds but alsoπ-π interactions could be successfully introduced in QAAR investigations. It is believed
that the QAAR studies based on aggregate models could be
applicable in other pesticide systems,which might facilitate the
discovery of improved compounds.
Acknowledgments
We thanks for the financial supports from National Key
Technology R&D Program of China (No. 2011BAE06B05),National Natural Science Foundation of China (No. 21172070),National High Technology Research Development Program of China (No.
2011AA10A207),National Basic Research Program of China (No.
2010CB126100) and the Fundamental Research Funds for the
Central Universities.
![]()
Table 1
Structures and bioactivity of sulfonylurea compounds.

Fig. 1. Homo-dimer-aggregate structure of compound 4 based onπ-π stacking interaction.

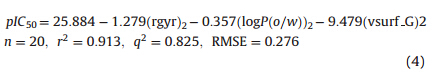
![]()
Table 2
Descriptors of monomer QSAR and aggregate QAAR models.

Fig. 2. Correlation plot of experimental activityversuspredicted activity,respectively for the monomer QSAR and aggregate QAAR models. (&) Values for compounds in the training set and (*) values for compounds in the test set.
{kind=link}
{kind=link}
| [1] | S.L. McGovern, E. Caselli, N. Grigorieff, B.K. Shoichet, A common mechanism underlying promiscuous inhibitors from virtual and high-throughput screening, J. Med. Chem. 45 (2002) 1712-1722. |
| [2] | J. Seidler, S.L. McGovern, T.N. Doman, B.K. Shoichet, Identification and prediction of promiscuous aggregating inhibitors among known drugs, J. Med. Chem. 46 (2003) 4477-4486. |
| [3] | S.C. Owen, A.K. Doak, P. Wassam, M.S. Shoichet, B.K. Shoichet, Colloidal aggregation affects the efficacy of anticancer drugs in cell culture, ACS Chem. Biol. 7 (2012) 1429-1435. |
| [4] | K.E.D. Coan, D.A. Maltby, A.L. Burlingame, B.K. Shoichet, Promiscuous aggregatebased inhibitors promote enzyme unfolding, J. Med. Chem. 52 (2009) 2067-2075. |
| [5] | Y.V. Frenkel, A.D. Clark Jr., K. Das, et al., Concentration and pH dependent aggregation of hydrophobic drug molecules and relevance to oral bioavailability, J. Med. Chem. 48 (2005) 1974-1983. |
| [6] | B. Vioque, J.M. Castellano, In vivo and in vitro 1-aminocyclopropane-1-carboxylic acid oxidase activity in pear fruit: role of ascorbate and inactivation during catalysis, J. Agric. Food Chem. 46 (1998) 1706-1711. |
| [7] | T. Yokozawa, E. Dong, T. Nakagawa, et al., In vitro and in vivo studies on the radical-scavenging activity of tea, J. Agric. Food Chem. 46 (1998) 2143-2150. |
| [8] | F. Fan, Z. Li, X.Y. Xu, X.H. Qian, Quantitative aggregation-activity relationship (QAAR): supermolecular view, dimer as the simplest aggregation state and monomolecule, QSAR Comb. Sci. 26 (2007) 737-743. |
| [9] | F. Fan, J.G. Cheng, Z. Li, X.Y. Xu, X.H. Qian, Novel dimer based descriptors with solvational computation for QSAR study of oxadiazoylbenzoyl-ureas as novel insect-growth regulators, J. Comput. Chem. 31 (2010) 586-591. |
| [10] | R.A. LaRossa, J.V. Schloss, The sulfonylurea herbicide sulfometuron methyl is an extremely potent and selective inhibitor of acetolactate synthase in Salmonella typhimurium, J. Biol. Chem. 259 (1984) 8753-8757. |
| [11] | G. Levitt, Synthesis and Chemistry of Agrochemicals II, American Chemical Society, Washington, 1991. |
| [12] | G.Z. Ye, Z.J. Fan, Z.M. Li, et al., Synthesis and herbicidal activity of new sulfonylurea derivatives, Chem. J. Chin. Univ. -Chin. 24 (2003) 1599-1603. |
| [13] | J.G. Wang, N. Ma, B.L. Wang, et al., Synthesis, crystal structure and biological activity of N-(4-Methyl-pyrimidin-2-yl)-N'-2-(nitrophenylsulfonyl)urea and its docking with yeast AHAS, Chin. J. Org. Chem. 26 (2006) 648-652. |
| [14] | B.L. Wang, N. Ma, J.G. Wang, et al., Synthesis and dimeric crystal structure of sulfonylurea compound N-[2-(4-methyl)pyrimidinyl]-N'-2-methoxycarbonylbenzene sulfonylurea, Chin. J. Struct. Chem. 23 (2004) 783-787. |
| [15] | J.G. Wang, Z.M. Li, N. Ma, et al., Structure-activity relationships for a new family of sulfonylurea herbicides, J. Comput. Aid. Mol. Des. 19 (2005) 801-820. |
| [16] | L. Pan, Z. Liu, Y.W. Chen, Y.H. Li, Z.M. Li, Design, synthesis and serbicidal activity of novel sulfonylureas containing monosubstituted pyrimidine moiety, Chem. J. Chin. Univ. 34 (2013) 1416-1422. |
| [17] | Y.W. He, C.W. Niu, X. Wen, Z. Xi, Molecular drug resistance prediction for acetohydroxyacid synthase mutants against chlorsulfuron using MB-QSAR, Chin. J. Chem. 31 (2013) 1171-1180. |
| [18] | K. Roy, S. Paul, Docking and 3D-QSAR studies of acetohydroxy acid synthase inhibitor sulfonylurea derivatives, J. Mol. Model. 16 (2010) 951-964. |
| [19] | M. Bitencourt, M.P. Freitas, MIA-QSAR evaluation of a series of sulfonylurea herbicides, Pest. Manag. Sci. 64 (2008) 800-807. |
| [20] | Z. Xi, Z.H. Yu, Z.W. Niu, S.R. Ban, G.F. Yang, Development of a general quantumchemical descriptor for steric effects: density functional theory based QSAR study of herbicidal sulfonylurea analogues, J. Comput. Chem. 27 (2006) 1571-1576. |
| [21] | S.R. Ban, C.W. Niu, W.B. Chen, et al., Interaction and CoMFA studies on A. thaliana acetohydroxyacid synthase by sulfonylureas, Chem. J. Chin. Univ. 28 (2007) 543-547. |
| [22] | J.L. Li, Y.C. Hang, C.Y. Geng, et al., QSAR studies on herbicidal activities of sulfonylurea compounds, Chem. J. Chin. Univ. 28 (2007) 539-542. |
| [23] | B.L. Wang, N. Ma, J.G. Wang, et al., 3D-QSAR analysis of new sulfonylureas related to their herbicidal activity, Acta Phys. -Chim. Sin. 20 (2004) 577-581. |
| [24] | G.F. Yang, H.Y. Liu, H.Z. Yang, QSAR and 3D-QSAR analysis of structurally diverse ALS inhibitors: sulfonylureas and triazolopyrimidine-2-sulfonamides, Pestic. Sci. 55 (1999) 1143-1150. |
| [25] | Z.H. Yu, C.W. Niu, S.R. Ban, X. Wen, Z. Xi, Study on structure-activity relationship of mutation-dependent herbicide resistance acetohydroxyacid synthase through 3D-QSAR and mutation, Chin. Sci. Bull. 52 (2007) 1929-1941. |
| [26] | Y. Ma, L. Jiang, Z.M. Li, C.M. Lai, 3D-QSAR study on N-(4-substituted pyrimidin-2-yl) benzyl sulfonylurea and phenoxy sulfonylurea, Chem. J. Chin. Univ. 25 (2004) 2031-2033. |
| [27] | X.H. Qian, Quantitative studies on structure-activity relationship of sulfonylurea and benzoylphenylurea type pesticides and their substituents' bioisosterism using synthons' activity contribution, J. Agric. Food Chem. 47 (1999) 4415-4418. |
| [28] | Z. Wang, J.J. Ye, R. Wu, et al., Bi-aryl rotation in phenyl-dihydroimidazoquinoline catalysts for kinetic resolution of arylalkyl carbinols, Catal. Sci. Technol. (2014), http://dx.doi.org/10.1039/C3CY00904A. |
| [29] | L. Ståhle, S. Wold, Multivariate data analysis and experimental design in biomedical research, Prog. Med. Chem. 25 (1988) 291-338. |
| [30] | J.J.P. Stewart, Optimization of parameters for semiempirical methods I. Method, J. Comput. Chem. 10 (1989) 209-220. |
| [31] | J.J.P. Stewart, Optimization of parameters for semiempirical methods II. Applications, J. Comput. Chem. 10 (1989) 221-264. |
| [32] | R. Todeschini, V. Consonni, Molecular Descriptors for Chemoinformatics, John Wiley & Sons, New York, 2009. |
| [33] | Y. Duan, P.A. Kollman, Pathways to a protein folding intermediate observed in a 1-microsecond simulation in aqueous solution, Science 282 (1998) 740-744. |
| [34] | T.J. Hou, K. Xia, W. Zhang, X.J. Xu, ADME evaluation in drug discovery. 4. Prediction of aqueous solubility based on atom contribution approach, J. Chem. Inf. Comp. Sci. 44 (2004) 266-275. |
| [35] | J. Sangster, Octanol-water partition coefficients: fundamentals and physical chemistry, Eur. J. Med. Chem. 32 (1997), 842. |
| [36] | T.J. Hou, X.J. Xu, ADME evaluation in drug discovery. 2. Prediction of partition coefficient by atom-additive approach based on atom-weighted solvent accessible surface areas, J. Chem. Inf. Comp. Sci. 43 (2003) 1058-1067. |
| [37] | G. Cruciani, P. Crivori, P.A. Carrupt, B. Testa, Molecular fields in quantitative structure-permeation relationships: the VolSurf approach, J. Mol. Struct. (Theochem.) 503 (2000) 17-30. |