




b Department of Chemistry, Nanjing University of Science and Technology, Nanjing 210094, China;
c Institute of Material Science and Engineer, Southwest University of Science and Technology, Mianyang 621010, China
1. Introduction
Energetic cocrystals are currently a major frontier in developing smart and novel energetic materials [1, 2, 3, 4]. Energetic-energetic cocrystals can not only provide intermediates for the production of new polymorphs but also improve the properties of existing energetic compounds [5, 6, 7]. Nowadays the study of energetic- energetic cocrystals is in its early stages,and only the teams in China and United States focus on the cocrystallization of energetic materials [7, 8, 9, 10]. Most reported energetic cocrystals were developed through screening different stoichiometries of existing compounds and there are no reliable strategies to rationally design energetic cocrystals. Kira B. Landenberger etc. [10] suggested that ‘‘an electron-rich energetic compound (rare among energetic compounds) with favorable geometry for interacting with electron-deficient rings (the properties of most energetic compounds) must be found’’. They also reported and analyzed two energetic molecules with electron-rich rings,diacetone diperoxide and triacetone triperoxide. However,they did not indicate how to quickly and efficiently search for the energetic materials with ‘‘electron-rich rings’’,which is the aim of the present paper.
Energetic eutectics have been reported since 1995 [11, 12, 13]. In the pharmaceutical field,the eutectics and cocrystals are often discussed together [14, 15],which has not been considered in the energetic material filed. Actually,by analyzing the reported papers in pharmaceuticals field [16, 17, 18, 19] and the phase diagram containing novel stable compounds,with at least two eutectic points was a prerequisite for two precursors to form a cocrystal (Table 1). Based on this discovery,we investigated the molecular structures, electronic structures and the Hansen solubility parameters (HSPs) [16] of 18 energetic molecules selected from the binary eutectic mixtures [20] to search for the smart precursors of energetic- energetic cocrystals.
| Table 1 Description of distinguishing characteristics of cocrystals (cc) from eutectic (eu) mixtures [16]. |

DMol3,the density functional theory (DFT) quantum mechanical code,was used to optimize the structures of the selected 18 energetic compounds. The exchange-correlation interaction was treated by functional Perdew,Burke and Ernzerh of generalized gradient approximation (PBE-TS GGA) [21],and applied basis set was double numerical basis set plus d-functions (DND). A convergence criterion of 10-6 a.u. on the total energy was used in the self-consistent field (SCF) calculations. The global orbital cutoff was taken to be 400 ppm. Minimization of the generated structure was performed using the convergence threshold. The value of the maximum energy change is 1 × 10-5 Hartree. The maximum force and maximum displacement are 0.02 Hartree/nm and 0.05 nm,respectively. The core treatment parameter was described by the all-electron method.
Prediction of HSPs was made using the Stefanis-Panayiotou group-contribution method [22]:

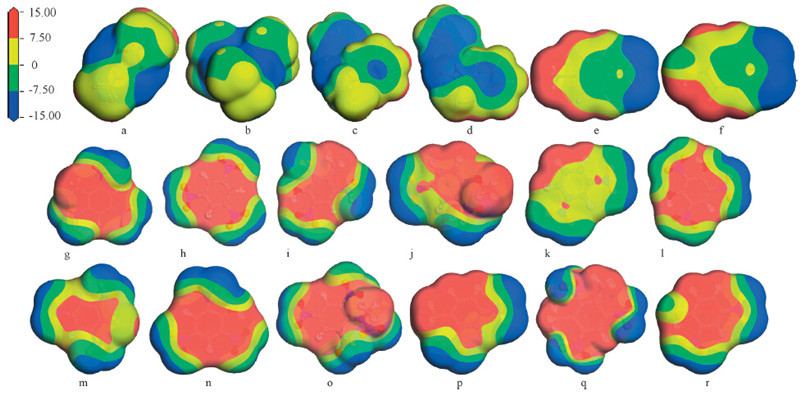
Fig. 1 shows the optimized structures of the 18 molecules (a-r). Based on the optimized structures,the electrostatic potential (ESP) surface of the 18molecules is obtained (Fig. 2). In Fig.2,it is clear that the positive electrostatic of the first six molecules (a-f) mainly appears in the periphery of the molecules. Accordingly,most of the negative electrostatic concentrates in the center of the molecules. And the ESP distribution of the remaining 12 molecules (g-r) is just opposite to that of the six precursors,which is consistent with the notion that most energetic molecules contain ‘‘electron-deficient rings or cages’’ in the molecule centers. The ‘‘electron-deficient rings or cages’’ should be caused by their electron-withdrawing nitro substitution. Then,we can infer that the first sixmoleculesmay be good for cocrystallizing with molecules g-r and many other energetic molecules with ‘‘electron-deficient rings or cages’’ by π-π intermolecular interactions. In addition,due to the larger electron cloud overlap,π-π intermolecular interactions are always stronger than p-π intermolecular interactions. It is known that p-π intermolecular interactions are the main interactions that maintain the stability of most reported energetic-energetic cocrystals [9, 23]. Actually,these cocrystals are often easy to turn into their precursors [9],which can be explained by the weak p-π intermolecular interactions. Finally,the inferred cocrystals in this paper with π-π intermolecular interactions will be more stable and worth further research.

|
Download:
|
| Fig. 1.The optimized structures of the 18 molecules (a,diacetone diperoxide (DADP); b,triacetone triperoxide (TATP); c,N,N'-diethyl-N,N'-diphenylurea; d,N,N'-dimethyl- N,N'-diphenylurea; e,4-nitrotoluene; f,4-nitrophenol; g,2,4,6-trinitrotoluene (TNT); h,1,3,5-trinitrobenzene (TNB); i,Trinitro-m-cresol (TNCr); j,2,4-dinitroanisole (DNAnisole); k,1,3-dinitrobenzene (DNB); l,styphnic acid (SA); m,picryl chloride (PkCl); n,picric acid (PA); o,Teryl; p,2,4-dinitrotoluene (DNT); q,trinitro-m-xylene (TNX); r, 2,4-dinitrophenol (DNPh)). | |

|
Download:
|
| Fig. 2.The electrostatic potential surface of the 18 molecules (red and blue surfaces represent electron poor and rich regions,respectively,with colors representing values between 15.00 kcal/mol and -15.00 kcal/mol) (a,DADP; b,TATP; c,N,N'-diethyl-N,N'-diphenylurea; d,N,N'-dimethyl-N,N'-diphenylurea; e,4-nitrotoluene; f,4-nitrophenol; g,TNT; h,TNB; i,TNCr; j,DNAnisole; k,DNB; l,SA; m,PkCl; n,PA; o,Teryl; p,DNT; q,TNX; r,DNPh). | |
Hansen solubility parameters (HSPs) can be used to indicate cocrystal formation and guide cocrystal screening,which is used widely in the pharmaceutical filed [20, 22]. In Table 2,it is notable that the values of these solution parameters are between -5.00 MP0.5 and 5.00 MP0.5 relative to those of TNT,which accords with the demand of miscibility [20]. The absolute value of the lowest unoccupied molecular orbital (LUMO) of the title four compounds are less than 3.00 eV,and the absolute value of the LUMO of the rest electron-deficient rings containing energetic compounds are all more than 3.00 eV. It indicates that the utilization of LUMO may be a good method to identify energetic compounds with electron-rich rings.
| Table 2 The molecular orbitals and solution parameters of 18 molecules. |

{kind=link}
{kind=link}
The results demonstrate that ESPs,HSPs and LUMO analyses can be used to search suitable precursors of cocrystals. Electronrich aromatic ring containing compounds such as 4-nitrotoluene, 4-nitrophenol,N,N'-dimethyl-N,N'-diphenylurea and N,N'-diethyl- N,N'-diphenylurea may be good candidate for developing novel smart energetic-energetic cocrystals. And more properties of these probable cocrystals,such as intermolecular interaction energy, sensitivity,detonation properties etc.,need to be studied. Additionally,with at least two eutectic points was a prerequisite for two precursors to form a cocrystal,which suggests that phase diagrams could be good guidance for developing smart cocrystals. Phase diagram investigation is underway.
AcknowledgmentsThe authors are grateful for financial support from National Natural Science Foundation of China - CAEP project (No. 11076002) and Science and Technology Found of CAEP (No. 2012A0302013).
| [1] | F. Lara-Ochoa, G. Espinosa-Pérez, Cocrystals definitions, Supramol. Chem. 19 (2007) 553-557. |
| [2] | N. Shan, M.J. Zaworotko, The role of cocrystals in pharmaceutical science, Drug Disc. Today 13 (2008) 440-446. |
| [3] | A.D. Bond, What is a co-crystal? CrystEngComm 9 (2007) 833-834. |
| [4] | P. Vishweshwar, J.A. Mcmahon, J.A. Bis, M.J. Zaworotko, Pharmaceutical cocrystals, J. Pharm. Sci. 95 (2006) 499-516. |
| [5] | L.E. Fried, M.R. Manaa, P.F. Pagoria, R.L. Simpson, Design and synthesis of energetic materials, Annu. Rev. Mater. Res. 31 (2001) 291-321. |
| [6] | J.P. Agrawal, R.D. Hodgson, Organic Chemistry of Explosives, Wiley, New York, 2007. |
| [7] | Z.W. Yang, H.Z. Li, X.Q. Zhou, et al., Characterization and properties of a novel energetic-energetic cocrystal explosive composed of HNIW and BTF, Cryst. Growth Des. 12 (2012) 5155-5158. |
| [8] | H.B. Zhang, C.Y. Guo, X.C. Wang, et al., Five energetic cocrystals of BTF by intermolecular hydrogen bond and π-stacking interactions, Cryst. Growth Des. 13 (2013) 679-687. |
| [9] | K.B. Landenberger, A.J. Matzger, Cocrystal engineering of a prototype energetic material: supramolecular chemistry of 2,4,6-trinitrotoluene, Cryst. Growth Des. 10 (2010) 5341-5347. |
| [10] | K.B. Landenberger, O. Bolton, A.J. Matzger, Two isostructural explosive cocrystals with significantly different thermodynamic stabilities, Angew. Chem. Int. Ed. 125 (2013) 6596-6599. |
| [11] | Z.R. Liu, Y.H. Shao, C.M. Yin, Y.H. Kong, Measurement of the eutectic composition and temperature of energetic materials. Part 1. The phase diagram of binary systems, Thermochim. Acta 250 (1995) 65-76. |
| [12] | C.M. Yin, Z.R. Liu, Y.H. Shao, Y.H. Kong, Measurement of the eutectic composition and temperature of energetic materials. Part 2. The HX-phase diagram of ternary systems, Thermochim. Acta 250 (1995) 77-83. |
| [13] | Y.H. Kong, Z.R. Liu, Y.H. Shao, C.M. Yin, W. He, Measurement of the eutectic composition and temperature of energetic materials. Part 3. The TX-phase diagram of ternary system, Thermochim. Acta 297 (1997) 161-168. |
| [14] | K. Chadwick, R. Davey, W. Cross, How does grinding produce co-crystals? Insights from the case of benzophenone and diphenylamine, CrystEngComm 9 (2007) 732-734. |
| [15] | E. Lu, H.N. Rodríguez, R. Suryanarayanan, A rapid thermal method for cocrystal screening, CrystEngComm 10 (2008) 665-668. |
| [16] | C.M. Hansen, The three-dimensional solubility parameter-key to paint component affinities: solvents, plasticizers, polymers, and resins. II. Dyes, emulsifiers, mutual solubility and compatibility, and pigments. Ⅲ. Independent calculation of the parameter components, J. Paint Technol. 39 (1967) 505-510. |
| [17] | R.E. Davis, K.A. Lorimer, M.A. Wilkowski, et al., Studies of phase relationships in cocrystal systems, ACA Trans. 39 (2004) 41-61. |
| [18] | R.D. Chapman, J.W. Fronabarger, A convenient correlation for prediction of binary eutectics involving organic explosives, Propellants Explos. Pyrotech. 23 (1998) 50-55. |
| [19] | D.J. Good, Ph.D., Pharmaceutical Cocrystal Eutectic Analysis: Study of Thermodynamic Stability, Solubility, and Phase Behavior, The University of Michigan, USA, 2010. |
| [20] | M.A. Mohammad, A. Alhalaweh, S.P. Velaga, Hansen solubility parameter as a tool to predict cocrystal formation, Int. J. Pharm. 407 (2011) 63-71. |
| [21] | Z.Y. Zheng, J.J. Zhao, Lattice energies and elastic properties of solid methane: assessment of different density functionals, Acta Phys. Chim. Sin. 28 (2012) 1809-1814. |
| [22] | E. Stefanis, C. Panayiotou, A new expanded solubility parameter approach, Int. J. Pharm. 426 (2012) 29-43. |
| [23] | H.R. Li, Y.J. Shu, S.J. Gao, et al., Easy methods to study the smart energetic TNT/CL- 20 co-crystal, J. Mol. Model. 19 (2013) 4909-4917. |