




b School of Science and Technology, Jiaozuo Teachers' College, Jiaozuo 454001, China
1. Introduction
Terminal alkynes are widely used in the fields of pharmaceuticals, agrochemicals, functional materials, and organic synthesis [1]. Their utilization in a wide range of cycloaddition [2] and coupling reactions [3] has stimulated a significant level of interest from chemists.
Terminal alkynes can be synthesized from carbonyl compounds via chain extension. The most frequently used reagents for converting aldehydes to terminal alkynes are CBr4/PPh3, CCl3CO2H/TsCl, and the Bestmann–Ohira reagent and its analogs, in which superbases, such as BuLi, NaHMDS, and t-BuOK are usually employed at low temperatures [4]. Acid chlorides can be converted to their corresponding alkynes when combined with a phosphorane reagent, followed by flash, vacuum pyrolysis at a high temperature of 750 ℃ [5]. Esters and Weinreb amides are also good substrates for this preparation, which undergo reduction, followed by a one-pot conversion to terminal alkynes [6]. Meanwhile, the dehydrobromination of 1- or 2-bromo-1-alkenes is a convenient method that has been developed in recent years [7]. In this procedure, both trans- and cis-configurations can be converted to terminal alkynes. The direct introduction of C≡CH residue into arenes and hetarenes through transition metalcatalyzed, cross-coupling reactions can generate the desired products. In this procedure, intermediate chemicals, such as acetylene, trimethylsilyl acetylene, propiolic acid, and ethynyltributyl stannane from suppliers, are often used [8]. In 2011, Huang found that the cleavage of 4-aryl-2-methyl-3-butyn-2-ols catalyzed by tetrabutylammonium hydroxide can produce terminal arylacetylenes [9]. This process is a rapid and efficient synthetic route, but the substrates are rare.
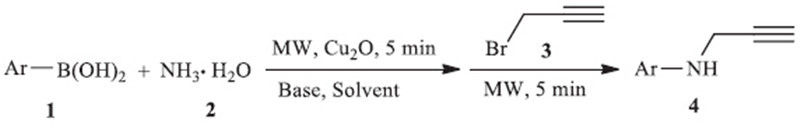
Enlightened by Fu’s green synthesis of primary aromatic amines by coupling aromatic boronic acids with aqueous ammonia [10], we here report a convenient and efficient microwave-assisted (MW), two-step synthesis of N-aryl propargylamine 4 via the coupling of aromatic boronic acid 1 with ammonia 2, and subsequent propargylation by propargyl bromide 3 in H2O (Scheme 1) as a green solvent.

|
Download:
|
| Scheme 1.One-pot synthesis of N-aryl propargylamine 4. | |
All the reactions were conducted using CEM Discover-SP microwave instrument. 1H NMR spectra were recorded using Bruker AM-500 and AM-400 spectrometer in CDCl3 with SiMe4 as an internal standard. IR spectra were performed on a Nexus FT-IR spectrophotometer. Commercially available reagents were used without further purification. All reactions were monitored by TLC with Huanghai GF254 silica gel-coated plates. Column chromatography was carried out using 300–400 mesh silica gel at medium pressure.
General procedure for the synthesis of 4: The microwave reaction tube was charged with boronic acid 1 (0.5 mmol), ammonia 2 (2 mmol, 25% aqueous solution), Cu2O (8 mg, 0.05 mmol), and H2O (2 mL). After the mixture was exposed to 5Wmicrowaves for 5 min, propargyl bromide 3 (59 mg, 0.5 mmol) was added. The mixture was then irradiated under 5W microwaves for another 5 min. The system was diluted with 30 mL of H2O after the reaction was completed, and the mixture was then extracted three times with EtOAc. The organic layer was separated, washed with water and saturated brine, and dried over anhydrous Na2SO4. The evaporation of the solvent provided the crude product, which was subjected to column chromatography (silica gel, EtOAcpetroleum ether 1:8–1:3) to yield N-aryl propargylamine 4.
4-Methyl-N-(prop-2-ynyl)aniline (4a) [11]: Yellow oil; 1H NMR (400 MHz, CDCl3): δ 7.03 (d, 2H, J = 8.18 Hz), 6.62 (d, 2H, J = 8.27 Hz), 3.91 (d, 2H, J = 2.22 Hz), 3.73 (s, 1H), 2.25 (s, 3H), 2.20 (s, 1H).
General procedure for the synthesis of 5: The tube was charged with AgSbF6 (17 mg, 0.05 mmol) after the propargylation process was completed. The mixture was irradiated by 5Wmicrowaves for 5 min. The system was diluted with 30 mL of H2O after completion of the reaction, and the mixture was then extracted with EtOAc three times. The organic layer was separated, washed with water and saturated brine, and dried over anhydrous Na2SO4. Evaporation of the solvent provided the crude product, which was then subjected to column chromatography (silica gel, EtOAc-petroleum ether 1:5–1:2) to obtain the quinoline derivatives 5.
6-Methylquinoline (5a) [12]: Light green oil; IR (KBr, cm-1): 3398, 3014, 1594, 1501, 1373, 1119, 829; 1H NMR (400 MHz, CDCl3): δ 8.85 (dd, 1H, J = 1.44, 4.12 Hz), 8.07 (d, 1H, J = 8.28 Hz), 8.00 (d, 1H, J = 8.6 Hz), 7.58 (m, 1H), 7.55 (dd, 1H, J = 1.88, 8.6 Hz), 7.37 (dd, 1H, J = 4.24, 8.28 Hz), 2.54 (s, 3H). 3. Results and discussion
The reaction of p-tolylboronic acid 1a (0.5 mmol), ammonia 2 (25% aqueous solution), propargyl bromide 3 (0.5 mmol), base (1 mmol), Cu2O, and 2 mL of solvent under microwave-assisted conditions was chosen as the model reaction for the preparation of the N-monopropargylated product, 4-methyl-N-(prop-2-ynyl)- aniline 4a, in which 5 min were allocated respectively in each step (Table 1).
| Table 1 Optimization of the synthesis to 4-methyl-N-(prop-2-ynyl)aniline 4a. |
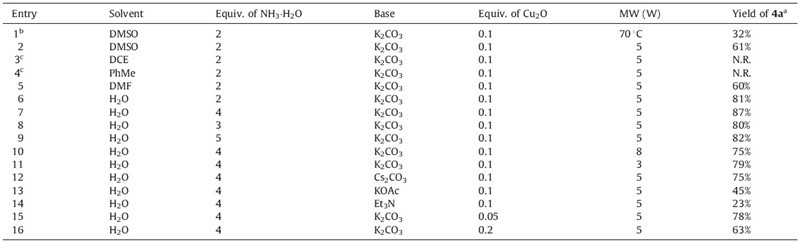
The realized yield of the product 1a was only 32% when the system was heated to 70 ℃ for 3 h in each step withoutMWenergy (Table 1, entry 1), but increased to 61% when a microwave power of 5W was used (entry 2). The reaction favored polar solvents, such as DMSO, DMF, and H2O, and satisfactory yields were observed (entries 2 and 5–16), whereas no product was detected when DCE, or PhMe, was used as the solvent (entries 3 and 4). The reaction can work smoothly in H2O, generating an excellent yield of 87% when 4 equiv. of ammonia and 0.1 equiv. of Cu2O were used (entry 7). A higher or lower loading of ammonia or Cu2O will decrease the yield (entries 8, 9, 15, and 16). The base, K2CO3, is more efficient than others, such as Cs2CO3, KOAc, and Et3N in this reaction (entries 11– 14). A microwave power of 5Wwas better than a lower, or higher, one (entries 7, 10, and 11). The reaction proceeded more efficiently when promoted by a microwave power of 5W in 2 mL of H2O for 5 min in each step respectively using p-tolylboronic acid 1a (0.5 mmol), ammonia 2 (4 equiv.), propargyl bromide 3 (0.5 mmol), Cu2O (0.1 equiv.), and K2CO3 as the base. The product, 4-methyl-N-(prop-2-ynyl)aniline 4a, was isolated with 87% yield when this optimum reaction condition was used (entry 7).
A series of aromatic boronic acid 1 were then subjected to this reaction under optimized reaction conditions. All the reactions were completed within the total 10 min, and moderate to excellent yields of N-aryl propargylamine 4 were achieved, as shown in Table 2. The reaction of aromatic boronic acids containing electrondonating groups, such as methyl and methoxyl, proceeded with higher yields (Table 2, 4a and 4c) compared to anilines containing electron-withdrawing groups, such as nitryl, bromo, chloro, and iodo (Table 2, 4d–4k). Notably, aromatic boronic acids containing either, an electron-donating, or an electron-withdrawing group at the para position can provide excellent yields (Table 2, 4a, 4c, 4d, 4h, and 4i). Although the substrates containing the o-substituent provided lower yields, the reactions can also be completed smoothly within total 10 min (Table 2, 4f and 4j).
| Table 2 Synthesis of N-aryl propargylamine 4.a |
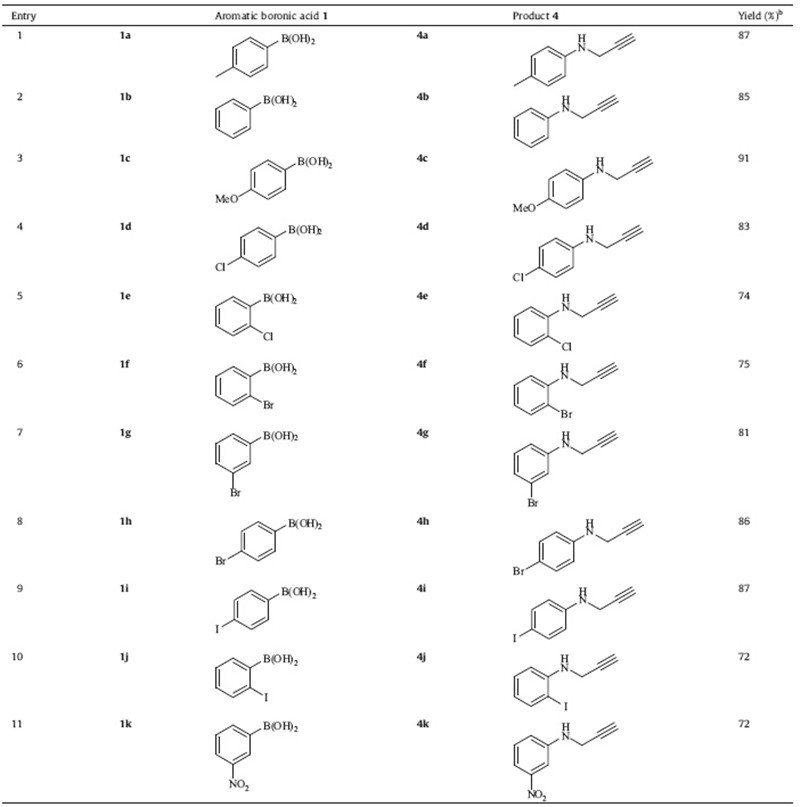
{kind=link}
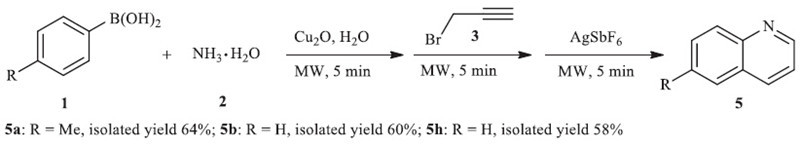
Moreover, this procedure can be used as a facile method for the synthesis of potential bioactive quinoline derivatives through a one-pot manipulation. Inspired by the report of Majumdar on silver-catalyzed cycloisomerization [12], we added a catalytic amount of AgSbF6 (0.1 equiv.) to the tube without isolating N-aryl propargylamine 4 when the two-step reaction was completed. The target molecule 5 can be isolated with good yields when the procedure was further conducted in 5W of microwave for an additional 5 min (Scheme 2).

|
Download:
|
| Scheme 2.One-pot synthesis of quinoline derivatives 5. | |
{kind=link}
In summary, we reported a one-pot microwave-assisted synthesis of N-aryl propargylamine. This method provides access via a quick and low-energy consumption process to terminal alkynes, which are important classes of intermediates in medicinal, chemical, and materials science.
AcknowledgmentsThe authors would like to thank the National Natural Science Foundation of China (No. 21262020), the Science and Technology Planning Project of Yunnan Province (No. KKSY201207140), the Natural Science Foundation of Yunnan Education Department (No. 09Y0081), and the Analysis and Measurement Foundation of Kunming University of Science and Technology (No. 20130560) for their financial supports.
| [1] | (a) J.M. Holub, K. Kirshenbaum, Tricks with clicks: modification of peptidomimetic oligomers via copper-catalyzed azide-alkyne [3+2] cycloaddition, Chem. Soc. Rev. 39 (2010) 1325-1337; (b) A. Soules, B. Ameduri, B. Boutevin, G. Calleja, Original fluorinated copolymers achieved by both azide/alkyne "click" reaction and Hay coupling from tetrafluoroethylene telomers, Macromolecules 43 (2010) 4489-4499; (c) Z.W. Chen, D.N. Ye, Y.P. Qian, M. Ye, L.X. Liu, Highly efficient AgBF4-catalyzed synthesis of methyl ketones from terminal alkynes, Tetrahedron 69 (2013) 6116- 6120; (d) S. Bew, G. Hiatt-Gipson, J.A. Lovell, C. Poullain, Mild reaction conditions for the terminal deuteration of alkynes, Org. Lett. 14 (2012) 456-459; (e) Z.M. Dong, Z.B. Ye, Synthesis of hyperbranched poly(phenylacetylene)s containing pendant alkyne groups by one-pot Pd-catalyzed copolymerization of phenylacetylene with diynes, Macromolecules 45 (2012) 5020-5031; (f) N. Onishi, M. Shiotsuki, T. Masuda, N. Sano, F. Sanda, Polymerization of phenylacetylenes using rhodium catalysts coordinated by norbornadiene linked to a phosphino or amino group, Organometallics 32 (2013) 846-853; (g) B.W. Zhou, H. Chen, C.Y. Wang, Mn-catalyzed aromatic C-H alkenylation with terminal alkynes, J. Am. Chem. Soc. 135 (2013) 1264-1267. |
| [2] | (a) A. Zarei, One-pot, efficient, and regioselective syntheses of 1,4-disubstituted 1,2,3-triazoles using aryldiazonium silica sulfates in water, Tetrahedron Lett. 53 (2012) 5176-5179; (b) W.S. Zhang, C.X. Kuang, Q. Yang, Synthesis of phenyl azides bearing (E)-2- halovinyl group, Res. Chem. Intermed. 38 (2012) 37-44; (c) Z.Z. Huang, R.L. Wang, S.R. Sheng, R.Y. Zhou, M.Z. Cai, Preparation of polystyrene- supported vinyl sulfone and its application in the solid-phase organic synthesis of 1-monosubstituted 1,2,3-triazoles, React. Funct. Polym. 73 (2013) 224-227; (d) Q. Yang, Y.B. Jiang, C.X. Kuang, Facile one-pot synthesis of monosubstituted 1- aryl-1H-1,2,3-triazoles from arylboronic acids and prop-2-ynoic acid (=propiolic acid) or calcium acetylide (=calcium carbide) as acetylene source, Helv. Chim. Acta 95 (2012) 448-454; (e) M. Xu, C.X. Kuang, Z. Wang, Q. Yang, Y.B. Jiang, A novel approach to 1- monosubstituted 1,2,3-triazoles by a click cycloaddition/decarboxylation process, Synthesis (2011) 223-228; (f) Y.B. Jiang, C.X. Kuang, Q. Yang, Facile and quick synthesis of 1-monosubstituted aryl 1,2,3-triazoles: a copper-free [3+2] cycloaddition, Tetrahedron 67 (2011) 289-292; (g) L. Wu, Y. Xie, Z. Chen, Y. Niu, Y. Liang, A convenient synthesis of 1-substituted 1,2,3-triazoles via CuI/Et3N catalyzed ‘click chemistry' from azides and acetylene gas, Synlett (2009) 1453-1456. |
| [3] | (a) Y.Y. Liu, C.P. Wang, X.B. Wang, J.P. Wan, Enaminone ligand-assisted homoand cross-coupling of terminal alkynes under mild conditions, Tetrahedron Lett. 54 (2013) 3953-3955; (b) D.H. Bai, C.J. Li, J. Li, X.S. Jia, New progress of acetylene-coupling reactions, Chin. J. Org. Chem. 32 (2012) 994-1009; (c) X.J. Niu, C.J. Li, J. Li, X.S. Jia, Importance of bases on the copper-catalyzed oxidative homocoupling of terminal alkynes to 1,4-disubstituted 1,3-diynes, Tetrahedron Lett. 53 (2012) 5559-5561; (d) L.L. Li, C.Y. Nan, Q. Peng, Y.D. Li, Selective synthesis of Cu2O nanocrystals as shape-dependent catalysts for oxidative arylation of phenylacetylene, Chem. Eur. J. 18 (2012) 10491-10496; (e) Z.Q. Weng, H.F. Li, W.M. He, et al., Mild copper-catalyzed trifluoromethylation of terminal alkynes using an electrophilic trifluoromethylating reagent, Tetrahedron 68 (2012) 2527-2531; (f) S.S. Patil, R.P. Jadhav, S.V. Patil, V.D. Bobade, Ligand and solvent-free iron catalyzed oxidative alkynylation of azoles with terminal alkynes, Tetrahedron Lett. 52 (2011) 5617-5619; (g) X.P. Nie, S.L. Liu, Y. Zong, P.P. Sun, J.C. Bao, Facile synthesis of substituted alkynes by nano-palladium catalyzed oxidative cross-coupling reaction of arylboronic acids with terminal alkynes, J. Organomet. Chem. 696 (2011) 1570-1573; (h) H. Xu, S.J. Gu, W.Z. Chen, D.C. Li, J.M. Dou, TBAF-mediated reactions of 1,1- dibromo-1-alkenes with thiols and amines and regioselective synthesis of 1,2- heterodisubstituted alkenes, J. Org. Chem. 76 (2011) 2448-2458; (i) B. Movassagh, M. Navidi, A simple and effective approach to the synthesis of alkynyl selenides from terminal alkynes, Chin. Chem. Lett. 23 (2012) 1035-1038; (j) Q.F. Zhou, X.P. Chu, S. Zhao, T. Lu, W.F. Tang, BF3·Et2O promoted conjugate addition of ethanethiol to electron-deficient alkynes, Chin. Chem. Lett. 23 (2012) 639-642; (k) M. Bakherad, A. Amin, A. Keivanloo, B. Bahramian, M. Raessi, Using Pd-salen complex as an efficient catalyst for the copper- and solvent-free coupling of acyl chlorides with terminal alkynes under aerobic conditions, Chin. Chem. Lett. 21 (2010) 656-660. |
| [4] | E. Quesada, S.A. Raw, M. Reid, E. Roman, R.J.K. Taylor, One-pot conversion of activated alcohols into 1,1-dibromoalkenes and terminal alkynes using tandem oxidation processes with manganese dioxide, Tetrahedron 62 (2006) 6673-6680. |
| [5] | R. Aitken, S. Seth, Convenient 2-step conversion of acid-chlorides to terminal alkynes, Synlett (1990) 211. |
| [6] | H.D. Dickson, S.C. Smith, K.W. Hinkle, A convenient scalable one-pot conversion of esters and Weinreb amides to terminal alkynes, Tetrahedron Lett. 45 (2004) 5597-5599. |
| [7] | (a) X.Z. Cheng, J. Jia, C.X. Kuang, Convenient synthesis of terminal alkynes from anti-3-aryl-2,3-dibromopropanoic acids using a K2CO3/DMSO system, Chin. J. Chem. 29 (2011) 2350-2354; (b) S. Shenawi-Khalil, S.U. Sonavane, Y. Sasson, Synthesis of acetylenes via dehydrobromination using solid anhydrous potassium phosphate as the base under phase-transfer conditions, Tetrahedron Lett. 53 (2012) 2295-2297; (c) M. Zhao, C.X. Kuang, Q. Yang, X.Z. Cheng, Cs2CO3-mediated synthesis of terminal alkynes from 1,1-dibromo-1-alkenes, Tetrahedron Lett. 52 (2011) 992-994. |
| [8] | (a) K. Park, G. Bae, J. Moon, et al., Synthesis of symmetrical and unsymmetrical diarylalkynes from propiolic acid using palladium-catalyzed decarboxylative coupling, J. Org. Chem. 75 (2010) 6244-6251; (b) K. Park, T. Palani, A. Pyo, S. Lee, Synthesis of aryl alkynyl carboxylic acids and aryl alrynes from propiolic acid and aryl halides by site selective coupling and decarboxylation, Tetrahedron Lett. 53 (2012) 733-737. |
| [9] | J. Li, P.C. Huang, A rapid and efficient synthetic route to terminal aryl-acetylenes by tetrabutylammonium hydroxide- and methanol-catalyzed cleavage of 4-aryl- 2-methyl-3-butyn-2-ols, Beilstein J. Org. Chem. 7 (2011) 426-431. |
| [10] | H.H. Rao, H. Fui, Y.Y. Jiang, Y.F. Zhao, Easy copper-catalyzed synthesis of primary aromatic amines by couplings aromatic boronic acids with aqueous ammonia at room temperature, Angew. Chem. Int. Ed. 48 (2009) 1114-1116. |
| [11] | M.A. Holman, N.M. Williamson, A.D. Ward, Preparation and cyclization of some N- (2,2-dimethylpropargyl) homo- and heteroaromatic amines and the synthesis of some pyrido[2,3-d]pyrimidines, Aust. J. Chem. 58 (2005) 368-374. |
| [12] | K.C. Majumdar, R.K. Nandi, S. Ganai, A. Taher, Regioselective synthesis of annulated quinoline and pyridine derivatives by silver-catalyzed 6-endo-dig cycloisomerization, Synlett 42 (2011) 116-120. |