




b School of Life Science, Northeast Agricultural University, Harbin 150030, China;
c Harbin FRP Institute, Harbin 150036, China;
d Zhejiang Hisun Pharmaceutical Co., Ltd., Taizhou 318000, China
1. Introduction
Vitamin D and its analogs constitute a valuable group of compounds that can be used to regulate gene expression in functions as varied as calcium and phosphate homeostasis as well as cell growth regulation and cell differentiation of a variety of cell types,such as keratinocytes,a type of epithelial cell [1]. Several such analogs have been developed as drugs on the market to cure patients suffering from osteoporosis,psoriasis,or renal dysfunction [2]. One of the analogs shown to exert a selective action on the parathyroid glands was 1α,25-dihydroxy-22-oxavitamin D3 (Maxacalcitol, 1),which was developed by Chugai pharmaceuticals in Japan and differs from 1,25(OH)2 D3 only by substitution of an oxygen in place of carbon 22 in the side chain [3]. It has been shown to be highly effective in stimulating monocytic differentiation of human promyelocytic leukemic HL-60 cells. Additionally, the clinical trials indicate that Maxacalcitol not only appears to be safe,suppressing PTH (secondary hyperparathyroidism) and exhibiting positive effects on bone formation,but is also less calcemic than 1,25(OH)2 D3 [4, 5]. The mechanism for the selectivity of Maxacalcitol can be attributed primarily to its low affinity for the serum vitamin D binding protein and altered pharmacokinetics [6]. To date,many methods has been described in the literature to synthesize Maxacalcitol [7, 8, 9, 10, 11],especially the strategy as shown in Scheme 1,which was employed by Chugai pharmaceuticals to prepare Maxacalcitol on the industrial scale. Scheme 1 starts with the ketodiol 3,which was prepared from dehydroepiandrosterone (DEA) 2 via microbiological 1a-hydroxylation [7, 8, 9]. Unfortunately,all of these processes suffer low yield (no more than 3%),and the starting material ketodiol 3 is monopolized by the company. Many ways for total synthesis of Maxacalcitol are only applicable in the laboratory. Additionally, the byproduct or isomer caused by the co-occurrence of side reactions is difficult to eliminate,leading to tedious work procedures and high production costs. Due to the lack of a more efficient synthetic route,the Maxacalcitol price remains high.

|
Download:
|
| Scheme 1.Chugai Pharmaceuticals reported synthetic scheme of Maxacalcitol. (a) Microbiological 1a-hydroxylation; (b) TBSCl,50-60 ℃; (c) NBS,and then γ-collidine, refluxing; (d) Ph3PEtBr,t-BuOK,r.t.; (e) 9-BBN,H2O2/NaOH,r.t.; (f) 4-bromo-1-butene,NaH; and then PdCl2,O2,r.t. (5); ethylacrylate,tetra-n-butylammonium hydroxide (6); N,N-dimethylacrylamide,NaH (7); (g) MeMgBr/MeCeCl2/MeLi; (h) hv,and then heat. | |
{kind=link}
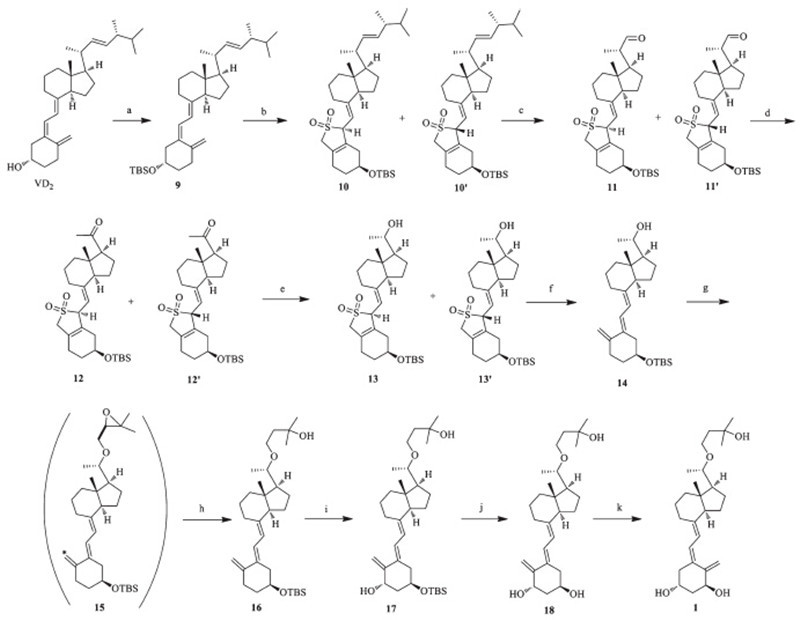
Herein,a novel and highly practical strategy for the synthesis of Maxacalcitol using ergocalciferol (vitamin D2) as the starting material and other readily available low cost raw materials and reagents has been developed. This method overcomes many weaknesses mentioned in the previous literature. The efficient and versatile synthesis will benefit those people suffering from hyperparathyroidism,dermatologic and psoriasis disease. 2. Experimental
3(S)-tert-Butyldimethylsilyloxy-9,10-secoergosta-5,7(E),10(19), 22(E)-tetraene (9): To a solution of ergocalciferol (200.0 g, 478mmol) in methylene chloride (1000 mL) under nitrogen, imidazole (7.8 g,115mmol) was added followed by TBSCl (8.6 g, 57.4mmol) at 0 ℃,and the mixture was stirred for 4 h at room temperature. The mixture was then treated with water (150mL), and the organic phase was separated,and the aqueous residue was extracted with methylene chloride (2× 100 mL). The combined solution was added into water (80 mL),and the mixture was used directly in the next step.
SO2-cycloadduct of 3(S)-tert-butyldimethylsilyloxy-9,10- secoergosta-5,7(E),10(19),22(E)-tetraene (10 or 10'): To the solution of TBS ether 9,SO2 flow was bubbled at room temperature and stirred for 8 h to produce a light green reaction mixture. The remaining sulfur dioxide and the methylene chloride solvent was removed under reduced pressure to afford the Diels-Alder adduct as a crude product. The residue was pumped under high vacuum to further remove SO2. The oil was taken directly to the next step.
SO2-cycloadduct of 3(S)-tert-butyldimethylsilyloxy-20(S)-formyl- 9,10-secoprega-5,7(E),10(19)-triene (11 or 11'): To the solution of SO2 adduct 10 in methylene chloride (600 mL),pyridine (7.8 g,115 mmol) was added,and O3 flow was bubbled at -65 ℃. The reaction mixture was stirred for 4 h until no more change was detected by TLC. This produced a light green reaction mixture. The reaction mixture was purged with air for 30 min,and the residue was concentrated under reduced pressure to afford 11 or 11' as an oil crude product. The product was taken directly to the next step.
SO2-cycloadduct of 3(S)-tert-butyldimethylsilyloxy-20-one- 9,10-secoprega-5,7(E),10(19)-triene (12 or 12'): 11 (50.7 g, 100 mmol) was stirred into dimethylformamide (500 mL). O2 was bubbled through the solution,and diazabicyclooctane (DABCO,11.2 g,100 mmol) was added,followed by addition of Cu(OAc)2 (3.6 g,20 mmol) and bipiridyl (3.1 g,20 mmol). The temperature was kept at a temperature of 35 ℃,and the solution was stirred at this temperature for 5 h. The reaction progress was checked by TLC. When the reaction was completed,ethyl acetate and water were added to the mixture. The aqueous phase was washed with ethyl acetate. The combined organic phases were washed with brine,dried over anhydrous magnesium sulfate,and filtered. Evaporation under reduced pressure at a temperature of 40 ℃ gave crude product 12 or 12' as a brown semi-solid,which was taken directly to the next step.
SO2-adduct of (3S)-tert-Butyldimethylsilyloxy-(20S)-hydroxy- 9,10-secopregna-5(Z),7(E),10(19)-triene (13 or 13'): 12 or 12' (49.2 g,100 mmol) was dissolved in dry THF (400 mL) and (R)-CBS (1 mol/L in THF,100 mL) was added dropwise slowly at -25 ℃, then BH3 was added dropwise (1 mol/L in THF,100 mL) at the same temperature. After being stirred for 1 h,saturated aqueous ammonium chloride solution (50 mL) was added,and the mixture was extracted with ethyl acetate. The combined organic phase was dried,filtered,and concentrated in vacuo to give light yellow oil that was used without purification in the next step.
13: [α]20D +58:0 (c 1.0,CH3Cl); 1H NMR (400 MHz,CDCl3): δ -0.01,-0.00 (d,6H),0.52 (s,3H),0.82 (s,9H),1.18-2.22 (m,20H), 2.52-2.56 (m,1H),3.56-3.68 (m,3H),3.91-3.94 (m,1H),4.55-4.58 (d,1H,J = 10.4 Hz),4.73-4.75 (d,1H,J = 10.4 Hz); 13C NMR (100 MHz,CDCl3): δ -4.7,-4.7,12.7,18.1,22.1,23.2,23.7,24.6, 24.7,25.8,29.3,29.7,31.0,34.1,39.1,44.6,55.9,58.2,58.5,66.8, 66.9,69.9,110.2,126.8,130.3,149.3; MS (ESI) m/z: 495 [M+H]+.
13':[α]20D +7:2 (c 1.0,CH3Cl); 1HNMR(400 MHz,CDCl3): δ -0.01, -0.00 (d,6H),0.60 (s,3H),0.82 (s,9H),1.17-2.20 (m,20H),2.54- 2.57 (m,1H),3.59 (s,2H),3.63-3.68 (m,1H),3.93-3.96 (m,1H), 4.44-4.47 (d,1H,J = 9.2 Hz),4.64-4.66 (d,1H,J = 9.2 Hz); 13C NMR (100 MHz,CDCl3): δ -4.7,12.4,18.1,22.0,23.6,24.3,25.0,25.8, 29.7,29.7,30.7,34.3,39.3,45.3,56.1,58.1,58.7,66.5,67.6,70.3, 110.8,128.5,130.7,150.0; MS (ESI) m/z: 495 [M+H]+.
(3S)-tert-Butyldimethylsilyloxy-(20S)-hydroxy-9,10-secopregna- 5,7(E),10(19)-triene (1413 or 13' was dissolved in ethanol (95%,400 mL) and NaHCO3 was added. The reaction mixture was warmed to reflux and stirred at this temperature for 3 h. When the reaction was completed,the mixture was filtered though a Buchner funnel and the solvent was evaporated under reduced pressure at 40 ℃. The product was purified on a chromatographic column using silica gel with mixtures of ethyl acetate in hexane,to give 35.5 g of 14 as colorless oil (yield 47% in six steps).
14: [α]20D +67:3 (c 1.0,CH3Cl); 1H NMR (400 MHz,CDCl3): δ -0.01 (d,6H),0.50 (s,3H),0.82 (s,9H),1.16 (d,3H,J = 6 Hz),1.18- 1.23 (m,2H),1.35-2.22 (m,13H),2.38-2.43 (m,1H),2.57-2.61 (m, 1H),2.79-2.83 (m,1H),3.64-3.67 (m,1H),3.78-3.81 (m,1H),4.58 (s,1H),4.86 (s,1H),5.81 (d,1H,J = 11.6 Hz),6.40 (d,1H, J = 11.6 Hz); 13C NMR (100 MHz,CDCl3): δ -4.7,-4.6,12.7,18.2, 22.2,23.2,23.6,25.0,25.9(3C),28.8,31.2,35.2,37.5,39.5,44.9, 56.3,58.7,69.4,70.3,107.5,116.5,119.9,136.6,142.9,150.0; MS (ESI) m/z: 431 [M+H]+; HRMS (ESI) m/z calcd. for C26H42O4: 431.3267 [M+H]+,found: 431.3253.
(3S)-tert-Butyldimethylsilyloxy-22-oxa-25-hydroxy-9,10- secopregna-5,7(E),10(19)-triene (16): To the secondary alcohol 14 (43.1 g,100 mmol) in THF (430 mL),NaH was added (60% dispersion in mineral oil,6 g,150 mmol) portionwise with stirring at room temperature for 0.5 h. After the evolution of hydrogen ceased,epoxy bromide (31 g,200 mmol) was added dropwise at the same temperature with stirring,and the mixture was refluxed for 5 h. After cooling to room temperature,lithium sec-butylborohydride (L-Selectride,1 mol/L in THF,200 mL,55.06 mmol) was added dropwise,and the mixture was stirred for 3 h. The mixture was then treated with saturated aqueous ammonium chloride solution (100 mL),the organic phase was separated,and the aqueous residue was extracted with ethyl acetate (2×100 mL). The combined extracts were washed with brine,dried with anhydrous magnesium sulfate,and evaporated under reduced pressure at 40 ℃. The product was purified on a chromatographic column using silica gel with mixtures of ethyl acetate in hexane to give 40.3 g of 16 as light yellow oil (yield 78%).
16: [α]20D +82:1 (c 1.0,CH3Cl); 1H NMR (400 MHz,CDCl3): δ -0.00 (s,3H),-0.06 (s,3H),0.48 (s,3H),0.82 (s,9H),0.72-0.97 (m, 2H),1.13 (d,3H,J = 6 Hz),1.17 (s,3H),1.18 (s,3H),1.19-1.27 (m, 2H),1.41-1.98 (m,12H),2.17-2.22(m,1H) 2.39-2.42 (m,1H), 2.56-2.61 (m,1H),2.78-2.82 (m,1H),3.17-3.21 (m,1H),3.41-3.44 (m,1H),3.77-3.81 (m,3H),4.58 (s,1H),4.86 (s,1H),5.80 (d,1H, J = 11.2 Hz),6.39 (d,1H,J = 11.6 Hz); 13C NMR (100 MHz,CDCl3): δ -4.7,-4.6,12.7,18.2,18.8,22.2,23.2,25.9(3C),26.0,28.8,29.1, 29.4,31.2,35.2,37.5,39.6,41.5,44.7,56.2,57.1,65.6,69.4,70.5, 79.0,107.6,116.5,119.9,136.5,142.8,150.0; MS (ESI) m/z: 517 [M+H]+.
(1S),(3R)-3-tert-Butyldimethylsilyloxy-22-oxa-1,25-dihydroxy- 9,10-secopregna-5,7(E),10(19)-triene (17): 16 (41.2 g,80 mmol) was dissolved in methylene chloride (500 mL),and NMO (18.7 g, 160 mmol) and SeO2 (4.4 g,40 mmol) were dissolved into the reaction. The reaction mixture was warmed to reflux and stirred at this temperature for 5 h. When the reaction was completed,the mixture was treated with water (250 mL),the organic phase was separated,and the aqueous residue was extracted with methylene chloride (100 mL). The methylene chloride phase was dried over anhydrous magnesium sulfate and filtered. The solvent was evaporated under reduced pressure at 38 ℃. The product was purified on a chromatographic column using silica gel with mixtures of ethyl acetate in hexane to give 20.4 g of 17 as colorless oil (yield 48%).
17: [α]20D +159:3 (c 1.0,CH3Cl); 1H NMR (400 MHz,CDCl3): δ -0.01 (s,6H),0.46 (s,3H),0.83 (s,9H),1.12 (d,3H,J = 6 Hz),1.16 (s, 3H),1.17 (s,3H),1.18-1.27 (m,2H),1.42-1.97 (m,14H),2.31-2.37 (m,1H),2.43-2.47 (m,1H),2.77-2.81 (m,1H),3.16-3.20 (m,1H), 3.41-3.44 (m,1H),3.75-3.80 (m,2H),4.11-4.14 (m,1H),4.41-4.44 (m,1H),4.88 (s,1H),4.99 (s,1H),5.78 (d,1H,J = 11.6 Hz),6.42 (d, 1H,J = 11.6 Hz); 13C NMR (100 MHz,CDCl3): δ -4.8,-4.7,12.6, 18.1,18.8,22.2,23.2,25.9(3C),26.0,28.9,29.1,29.4,37.0,39.6, 41.5,42.9,44.8,56.2,57.1,65.6,66.8,70.5,70.6,79.0,107.7,116.6, 122.2,134.6,143.3,153.1; MS (ESI) m/z: 555 [M+Na]+.
1(S),(3R)-1,3,25-Trihydroxy-9,10-secopregna-5,7(E),10(19)-triene (18): 17 (26.6 g,50 mmol) and tetrabutyl ammonium fluoride (19.5 g,75 mmol) were dissolved in THF (270 mL),and the reaction mixture was refluxed for 8 h. After cooling to room temperature the organic layer was evaporated under reduced pressure at 40 ℃. The residue was dissolved in ethyl acetate (150 mL),transferred to a separatory funnel,and washed with water (80 mL) and saturated sodium chloride solution (80 mL). The ethyl acetate phase was dried over anhydrous magnesium sulfate and filtered,and the evaporation afforded the crude oil carbinol 18. The product was purified on a chromatographic column using silica gel with mixtures of ethyl acetate in hexane to give 18.0 g of 18 as a colorless oil (yield 86%). 18: [α]20D +168:2 (c 1.0,MeOH); 1H NMR (400 MHz,CDCl3): δ 0.54 (s,3H),1.19 (s,3H,J = 5.6 Hz),1.23 (s,6H),1.24-1.37 (m,2H), 1.48-2.08 (m,13H,),2.24-2.30 (m,1H),2.55(s,br,1H),2.65 (s,br, 1H),2.81-2.88 (m,2H),3.24-3.27 (m,1H),3.46-3.51 (m,1H), 3.82-3.90 (m,2H),4.19-4.23 (m,1H),4.47-4.49 (m,1H),4.96 (s, 1H),5.10 (s,1H),5.89 (d,1H,J = 11.2 Hz),6.55 (d,1H,J = 11.2 Hz); 13C NMR (100 MHz,CDCl3): δ 12.8,18.9,22.2,23.2,25.8,28.9,29.1, 29.2,38.7,39.5,41.5,41.9,44.8,56.2,57.1,65.5,65.6,70.7,70.8, 78.9,109.5,116.5,122.8,133.5,144.0,151.8; MS (ESI) m/z: 441 [M+Na]+; HRMS (ESI) m/z calcd. for C26H42O4: 441.2981 [M+Na]+, found: 441.2928; tR = 8.52 min.
1α,25-Dihydroxy-22-oxavitamin D3 (1): The triol 18 (21 g, 50 mmol) and 9-acetyl anthracene (2.1 g,10 mmol) were dissolved in acetone (3 L). The mixture was stirred and irradiated in a quartz apparatus equipped with a high-pressure mercury lamp at 0-5 ℃ for about 1 h. The reaction progress was checked by HPLC (99.8%), then evaporated under reduced pressure at 32 ℃. The product was purified on a chromatographic column using silica gel with mixtures of ethyl acetate in hexane (1:1) to give 19.3 g of 1 as a white solid (yield 92%).
1: Mp: 122-124 ℃; [α]20D +37:9 (c 1.0,CH3Cl); 1H NMR (400 MHz,DMSO-d6): δ 0.49 (s,3H),1.08 (s,6H),1.09 (d,3H, J = 1.6 Hz),1.22-1.28 (m,1H),1.39-1.65 (m,10H),1.79-1.84 (m, 3H),1.93-1.99 (m,1H),2.15-2.20 (m,1H),2.35-2.37 (m,1H), 2.78-2.81 (m,1H),3.18-3.21 (m,1H),3.25-3.31 (m,1H),3.60(q, 1H,J = 7.6 Hz),3.99-4.04 (m,1H),4.12(s,1H),4.18-4.21 (m,1H), 4.54 (d,1H,J = 4 Hz),4.76 (s,1H),4.86 (d,1H,J = 4.4 Hz),5.23 (s, 1H),5.99 (d,1H,J = 11.2 Hz),6.18 (d,1H J = 11.2 Hz); 13C NMR (100 MHz,DMSO-d6): δ 12.3,19.1,21.8,22.9,24.7,28.3,29.6,29.7, 38.9,43.1,43.2,44.1,44.9,55.5,56.8,64.3,65.1,68.2,68.4,76.7, 109.8,117.8,122.4,135.9,139.6,149.5; MS (ESI) m/z: 441 [M+Na]+; HRMS (ESI) m/z calcd. for C26H42O4: 441.2981 [M+Na]+,found: 441.2945; tR = 8.45 min. 3. Results and discussion
The synthetic strategy for the stereoselective synthesis of 1 starts from commercially available ergocalciferol. The route involves modification of the side chain present in the ring vitamin D2 precursor to a 1S-formylethyl group followed by elaboration of the new side chain present in the particular target compound. The synthesis the rest of the full vitamin D skeleton must be elaborated. We knew oxygen and light will destroy the sensitive triene system. The synthesis of 11 or 11' employed a modification of Calverley’s procedure and the new side chain in question [12].
In our present work (Scheme 2),the hydroxyl group in ring A of ergocalciferol was protected with a TBS group with TBSCl and imidazole in dichloromethane. Due to the instability of the triene moiety of the vitamin D nucleus,we prepared epimeric SO2 adducts 10 or 10' through Diels-Alder reaction by bubbling SO2 gas into the dual-solvent system of water and dichloromethane (1:4) at room temperature. Then the resulting sulfone was ozonized in dichloromethane with pyridine at -65 ℃ to give the aldehydes 11 or 11'. In each of the above reactions,the mixture was washed with water,the aqueous layer was extracted with dichloromethane,and the combined organic phase was dried over anhydrous magnesium sulfate,filtered,and concentrated to afford colorless or light yellow oils. An oxygenation procedure was taken to produce isomers 12 and 12' [13]. Although the methyl ketone reduction of 12 or 12' treated with NaBH4 could yield 13,the products were isomers with the undesired R type as the major part [14]. In order to obtain the desired 20(S)-alcohol 13 or 13' with high purity,an asymmetric synthetic strategy was employed. 13 or 13' was introduced by the (R)-MeCBS catalyzed asymmetric carbonyl reduction of ketone 12 or 12' using borane tetrahydrofuran complex (1 mol/L THF) as the reducing agent [15]. After the extrusion of SO2,the important intermediate 20(S)-alcohol 14 was afforded. Due to the fact that the epoxide exhibits high reactivity toward primary and secondary alcohols under Williamson conditions to give the corresponding ether,3-(bromomethyl)-2,2-dimethyloxirane was chosen as the alkylating agent to react with the secondary alcohol 14 to afford the epoxy-ether 15,which subsequently reacted with the boron reagents to give tertiary alcohol 16 as the major product [16]. Alcohol 16 was produced in one pot from 14 with lithium secbutylborohydride( L-Selectride) in THF at room temperature [17].

|
Download:
|
| Scheme 2.Reagents and conditions: (a) TBSCl,imidazol,DCM,0 ℃,4 h; (b) SO2,r.t.,8 h; (c) O3,pyridine,DCM,-65 ℃,4 h; (d) O2,DABCO,Cu(OAc)2,bipiridyl,DMF,5 h,35 ℃; (e) BH3,R-CBS,THF,-25 ℃,2 h; (f) NaHCO3,ethanol (95%),refluxed; (g) NaH,3-(bromomethyl)-2,2-dimethyloxirane,THF,refluxed; (h) L-selectride,THF,r.t.,3 h; (i) SeO2, NMO,DCM,refluxed; (j) Bu4NF,THF,refluxed,8 h; (k) acetone,9-acetyl anthracene,0 ℃,hv,1 h. | |
{kind=link}
16 was oxidized at C1 position with selenium dioxide and Nmethylmorpholine- N-oxide (NMO) under reflux in methylene chloride to produce 17 [18]. The subsequent desilylation of 17 with tetrabutylammonium fluoride yielded 18 in THF. Triplet-sensitized photoisomerization is well-known for the conversion of trans-vitamin D compounds to cis-vitamin D compounds [19],and the photoisomerization of 18 afforded the target Maxacalcitol 1. This reaction was performed in acetone at 0-5 ℃ and irradiated with a mercury light source in a quartz vessel using 9- acetylanthracene as a sensitizer. The crude photolysis reaction mixture was concentrated under reduced pressure and purified by silica gel column chromatography to afford Maxacalcitol 1 as a white solid with a yield of 92%. 4. Conclusions
In summary,a novel and efficient synthesis of Maxacalcitol with improved overall yield was developed. The asymmetric synthesis of key intermediate 14 was achieved,and the strategy of using SO2 to protect the sensitive triene system of the vitamin D skeleton significantly promoted the yield of Maxacalcitol. Therefore, the simple operation procedure and the low production costs of this developed synthetic route will benefit the industrial production of Maxacalcitol.
| [1] | G. Jones, Vitamin D analogs, Endocrin. Metab. Clin. 39 (2010) 447-472. |
| [2] | G.H. Posner, M. Kahraman, Organic chemistry of vitamin D analogues, Eur. J. Org. Chem. 24 (2003) 3889-3895. |
| [3] | A.J. Brown, Vitamin D analogs for the treatment of secondary hyperparathyroidism in chronic renal failure, in: T. Naveh-Many (Ed.), Molecular Biology of the Parathyroid, Kluwer Academic/Plenum Publishers, Austin, USA, 2005, pp. 95-112. |
| [4] | Y. Nishii, T. Okano, History of the development of new vitamin D analogs: studies on 22-oxacalcitriol (OCT) and 2β-(3-hydroxypropoxy) calcitriol (ED-71), Steroids 66 (2001) 137-146. |
| [5] | N.C. Harvey, K. Javaid, N. Bishop, et al., MAVIDOS maternal vitamin D osteoporosis study: study protocol for a randomized controlled trial. The MAVIDOS study group, Trials 13 (2012) 13. |
| [6] | A.J. Brown, E. Slatopolsky, Vitamin D analogs: therapeutic applications and mechanisms for selectivity, Mol. Aspects Med. 29 (2008) 433-452. |
| [7] | S. Hatakeyama, T. Okano, J. Maeyama, et al., Synthesis and evaluation of 1a 25- dihydroxy-22-oxavitamin D3(OCT), Bioorg. Med. Chem. 9 (2001) 403-415. |
| [8] | Y. Fall, V. González, B. Vidal, et al., Stereoselective synthesis of 22-oxacalcitriol (OCT) and analogues modified at C25, Tetrahedron Lett. 43 (2002) 427-429. |
| [9] | E. Murayama, K. Miyamoto, N. Kubodera, et al., Synthetic studies of vitamin D3 analogs VⅢ: synthesis of 22-oxavitamin D3 analogs, Chem. Pharm. Bull. (Tokyo) 34 (1986) 4410-4413. |
| [10] | N. Kubodera, H. Watanabe, T. Kawanishi, et al., Synthetic studies of vitamin D analogues XI: synthesis and differentiation-inducing activity of 1α, 25-dihydroxy- 22-oxavitamin D3 analogues, Chem. Pharm. Bull. (Tokyo) 40 (1992) 1494-1499. |
| [11] | T. Mikami, T. Iwaoka, M. Kato, et al., Practical formation of γ-keto or γ-hydroxy ether linkage from alcohol, Synth. Commun. 27 (1997) 2363-2369. |
| [12] | M.J. Calverley, Synthesis of MC 903 a biologically active vitamin D metabolite analogue, Tetrahedron 43 (1987) 4609-4619. |
| [13] | K.L. Perlman, H.M. Darwish, H.F. Deluca, 20-Oxopregnacalciferols: vitamin D compounds that bind the progesterone receptor, Tetrahedron Lett. 35 (1994) 2295-2298. |
| [14] | S. Anchel, W. Koby, R.G. Michal, et al., Polymorphs of Maxacalcitol and process for the preparation of Maxacalcitol, WO 2012/122451 A2 (2012). |
| [15] | R. Rodriguez, A.S. Chapelon, C. Ollivier, et al., Stereoselective synthesis of CD-ring precursors of vitamin D derivatives, Tetrahedron 65 (2009) 7001-7015. |
| [16] | H. Shimizu, K. Shimizu, N. Kuboru, et al., Efficient modification of steroid 20Shydroxy functionality for industrial preparation of 1a,25-dihydroxy-22-oxavitamin D3, Maxacalcitol, Tetrahedron Lett. 45 (2004) 1347-1350. |
| [17] | H. Shimizu, K. Shimizu, N. Kubodera, et al., Industrial synthesis of Maxacalcitol, the antihyperparathyroidism and antipsoriatic vitamin D3 analogue exhibiting low calcemic activity, Org. Process Res. Dev. 9 (2005) 278-287. |
| [18] | B.G. Anderson, W.E. Bauta, W.R. Cantrell, Development of an improved process for doxercalciferol via a continuous photochemical reaction, Org. Process Res. Dev. 16 (2012) 967-975. |
| [19] | L.D. Coutts, W.B. Geiss, B.T. Gregg, et al., A stereospecific synthesis of 24(S)- hydroxyvitamin D2, a prodrug for 1a,24(S)-dihydroxyvitamin D2, Org. Process Res. Dev. 6 (2002) 246-255. |