




b Department of Physical Sciences, Nicholls State University, PO Box 2022, Thibodaux, LA 70310, USA
Photochromic molecules have been widely used in a variety of new techniques and processes,such as sunglasses,optical switching,optical date storage,nonlinear optical devices,optical nanoparticles,and photonic crystal because of their controllable light-induced change of color [1, 2]. Among these molecules,a suitable spiropyran (SP) leading to a fast bidirectional photoswitching processes is one of the very recent most studied molecules [3, 4, 5, 6].
In Spiro-type compounds,the indole and benzopyran planar heterocycles are joined orthogonally by a common spiro carbon atom,which constrains the conjugation of the two p-electronic systems so that the lowest electronic transition occurs in the near UV region (l < 400 nm). Correspondingly,this molecule is colorless. After this excitation,the break of the bond between the Spiro-C and O atoms takes place and this spiropyran converts to a planar form in a few picoseconds [7]. This form is an open photoisomer of spiropyran,called merocyanine (MC) with absorption in the visible region (l = 500-700 nm) (see Scheme 1),since the planar conjugation leads to p-electronic delocalization. This process corresponds to a photocoloration reaction. As a photoreversible and thermal reaction,MC can switch back to the closed form SP by heating or irradiation in the visible region. The reversible process,accompanied by remarkable changes in geometries and charge distribution,can be used in various applications.
Thus,the photochromic reactions of SP have been widely studied by time resolved spectroscopy to find the complex mechanism [8, 9, 10, 11, 12]. In this paper,we only pay attention to the photo induced ring-opening reaction of BIPS,which is the primary process of SP,although several reactions exist with different substituents on the SP and experimental conditions. Recent spectroscopic investigations reveal the following aspects: (1) The efficient internal conversion from the excited state S1 to the ground state S0 leads to a ring-opening process to form photoproducts (MC),corresponding to quantum yields less than 10% in gas phase [9] and 3.3% in water [12]; (2) The MC products can not be directly produced from the excited SP,and some intermediate species/state are assumed in the ring-opening process [9]; (3) The formation of MC is likely to be barrierless and generally fast in 0.9 ps [7] or 1.6 ps [12],measured by femtosecond absorption spectroscopy or subpicosecond transient spectroscopy in water,respectively.
To clarify the mechanisms of the photo triggered ring-opening reactions of SP,various high level quantum calculations have been carried out,including time-dependent density functional theory (TD-DFT) [13],the complete active space self-consistent field (CASSCF) [14],complete active space second-order perturbation theory (CASPT2) [15] level,density matrix renormalization group (DMRG) based CASSCF,CASPT2 [16] from simplified [17] to more [18] and even more [19] realistic models of SP,and other approximations [13, 20]. The primary theoretical investigations have proposed three different pathways for the photochromic reaction of the electronically excited SP. All three pathways rely on the internal conversion through conical intersections. The first and second kinds of paths are both associated with the Cspiro-O stretching and hydrogen-out-of-plane (HOOP) torsion modes (the C atom is connected by Spiro-C) of the excited SP. The first route starts from the excitation of one isomer of SP (trans-SP),which transitions first to TCC(S1),which in turn forms CIS1/S0(TCC),which can transition to MC intermediates or back to the original SP molecule [18, 19]. Alternatively,cis-SP can become CCC(S1) after photo initialization,then decaying to CIS1/S0(CCC) and finally either MC isomers or the ground state SP,as reported by Liu and Morokuma [19]. The very recent theoretical study implies that these two channels involve not only a one-bond-flipping (OBF) path on the S1 state,but also a HOOP valley following the Cspiro-O stretching and H-C out-of-plane torsion modes as mentioned above [19]. The last pathway involves the lengthening Cspiro-N bond and the internal conversion from the electronically excited states to the SP ground state through CIS1/S0(CN) [18, 19]. All of these pathways play an important role in the effective internal conversion yields of SP.
Theoretical investigations on the photochromic reaction of SP typically use static quantum calculations with mode compounds to locate energy minima on the potential energy surfaces and deriving the minimum energy path by connecting the critical points. These calculations are fundamental and important in understanding the main features of the reaction mechanisms. However,the static calculations alone can not provide detailed information for a specific reaction path that is actually realized in a photochromic reaction due to the following limitations: (1) the simplified model can not represent the real molecular structures and conformational adaptability; (2) they are incapable of describing the time-dependent properties of the ring-opening process; (3) they are usually performed along one or two reaction coordinates with the other coordinates fixed or optimized and consequently are insufficient in characterizing a real photochromic process where all reaction coordinates are involved and play a nonnegligible role; and (4) they do not include the laser pulse properties and subsequently cannot provide detailed explanations for photochemical experiments in which the laser pulse parameters strongly influence the reaction.
To more directly interpret the mechanisms of this reaction,in this work,we study the dynamics of the radiationless deactivation, which is necessary for the photo-induced chemical reaction [21] of the low-lying excited states of SP following the excitation using several laser pulses,by a semiclassical dynamics simulation approach. In our simulation study,all degrees of freedom of the molecule are included in the calculations and the laser pulse is explicitly coupled to electrons. By monitoring the nuclear motions triggered by a specific laser pulse,we are able to describe a realistic photo ring-opening reaction path. The more accurate potential energy curves (PECs) of the ground state and the low-lying excited states along the reaction path will be supplied for the explanation of the simulation results. 2. Methodology
In our semiclassical dynamics simulation approach,called semiclassical electron-radiation-ion dynamics (SERID),the timedependent quantum states are calculated for the valence electrons, while the radiation field and the motion of the nuclei are treated classically.
The details of this method have been described elsewhere [22, 23],so only a brief outline is present here. The radiation field is directly introduced in the electronic Hamiltonian though a vector potential A,and the time-dependent Peierls substitution is used to couple the radiation field and electrons [24].
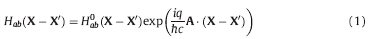
Here Hab(X - X') is the Hamiltonian matrix element for basis functions a and b on atoms at X and X0 respectively,and q = -e is the charge of the electron. The one-electron wave function is updated at each time step by solving the time-dependent Schro¨ dinger equation with an algorithm based on the timeevolution operator and conservation of probability [25]. Densityfunctional- based tight-binding (DFTB) is employed to calculate the electronic structure,in which only the valence electrons are treated and the remaining electrons plus nucleus are represented as a core [26].
The nuclear motion is treated classically and described by the Ehrenfest’s theorem [22, 23]
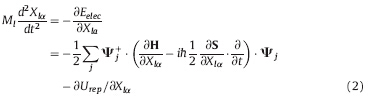
where Xlα = The ground state geometries of trans-SP conformerwas obtained
within 5000 fs of simulation. The primary dihedral angles N-C1-C3-
C6,C1-C3-C6-C9,C3-C6-C9-C12,and H21-C3-C1-C6 (see Scheme 1)
arehereafter labeledasa,b,gandt,respectively,consistentwiththe
previous work [19].
To produce an excited state,in which the photochromic ringopening
process of SP is appreciably activated,an 80 fs (FWHM)
laser pulse was applied to trans-SP conformer with an effective
photon energy of 3.972 eV,corresponding to 312.1 nm light,which
is in agreement with the mentioned experimental conditions [9].
The energy selected matches the energy gap between the
HOMO - 1 and LUMO + 1 in the present density-functional based
approach for the equilibrated ground state geometry. Simulations
were run for more than 100 different fluences,corresponding to
several initial conditions,but only representative results will be
presented for a fluence of 0.139 kJ/m2 that generates the desired
vibrational motions necessary for the ring-opening reaction.
Along the typical trajectory,the potential energy curves (PECs)
of the ground state and the low-lying two excited states are
constructed at the three state average (SA3)-CASSCF and the
following CASPT2 level with a level shift 0.1 of a.u. and the 6-
31G(d) basis set employing the MOLCAS 7.5 quantum chemistry
package [28]. The active space is constructed with 12 electrons
distributed in 10 molecular orbitals,consistent with the previous
calculations [19].
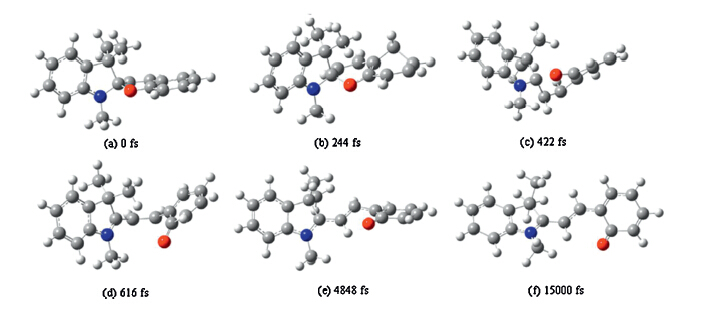
Six snapshots from the simulation at different times for the
photo induced ring-opening process of trans-SP are shown in Fig. 1.
The laser excitation starts at 0 fs and the initial molecule becomes
electronically excited after about 50 fs. The most impressive
features shown in Fig. 1 are the lengthening of the C1-O bond,the
twist around C3-C6 bond,and the HOOP torsion of phenyl ring
connected to the O atom,which is shown by the dihedral angle
H27-C13-C9-C15,denoted by θ.
The important nuclear motions for ring-opening reaction of SP,
described by the variations of C1-O bond and dihedral angles a,b,g
are presented in Fig. 2a and b,respectively. The evident variations
of these nuclear motions start at 422 fs. After this time,it can be
seen in Fig. 2a that the C1-O bond is gradually lengthened from the
initial 1.404Å to 3.618Å at 616 fs,and then the bond length
oscillates around an average value 3.5Å until about 4848 fs,when
this bond is broken and the six membered ring with the O atom is
accordingly opened. This bond is again elongated to 3.945Å at this
moment. After that,an average value of about 4.000Å is kept until
the end of this simulation. Correspondingly,Fig. 2b shows that the
dihedral angle a increases rapidly to the peak value 272.68 at 616 fs
and then decrease gradually to 158.48 at 4848 fs. After that,this
twist angle maintains an average value of 180.08 to the end. The
dihedral angles b and g are also speedily enlarged after 422 fs and
their values are basically kept until 4848 fs. At this moment,the
angle b increases to 180.48,while the angle g decreases to -37.78.
Similar with the angle a,the angles b and g then respectively retain
average values of 180.08 and 0.08 up to the end,respectively. With
these results,it can be indicated that the initial MC conformer at
the beginning of the ring-opening is MC-TCC. After that,the
transient MC-CCC form appears at 616 fs. Then,this isomer is
gradually switched back to the MC-TCC conformer. After 4848 fs,
MC-TCC isomerizes to the final form MC-TTC,which is the most
stable isomer of MC as observed previously by experimental
observations [8, 9, 10, 11, 12]. In fact,the molecular vibration is initialized
from two cooperative HOOP distortions of two connected six
membered rings,corresponding to the changes of dihedral angles t
and u as presented in Fig. 2c. These two dihedral angles vary visibly
from the beginning until about 422 fs. Then,they are changed back
to their initial values with large oscillations up to the end. These
HOOP distortions imply the presence of HOOP valleys as proposed
very recently [19].
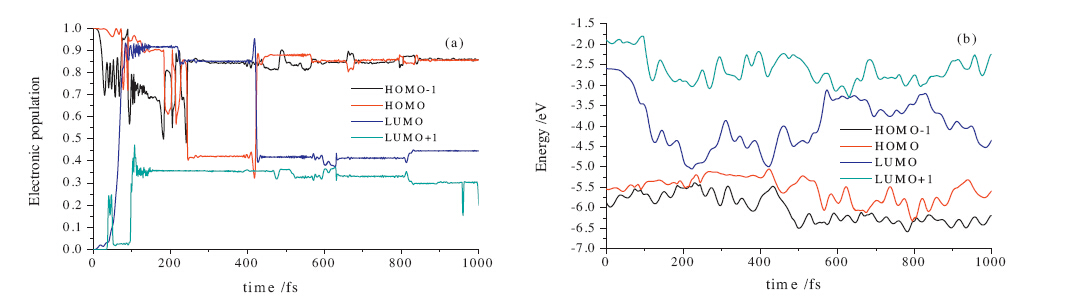
The populations and energies of HOMO - 1,HOMO,LUMO and
LUMO + 1 for this reaction path are also shown in Fig. 3a and b,
respectively. It can be seen in Fig. 3a that the electronic population
of HOMO - 1 decreases while that of LUMO increases from the
initial photon excitation until 244 fs. When the energy gap
between HOMO and LUMO is diminished according to the sudden
descent of LUMO energy and slight ascent of HOMO energy,then
the gap is enlarged till 422 fs. At this moment,these two orbitals
are again degenerate,corresponding to the fact that the electronic
population of LUMO decreases while that of HOMO increases. After
that,no remarkable change of the electron population occurs
between these four interesting frontier molecular orbitals.
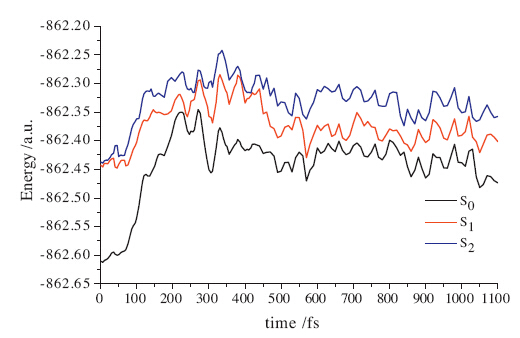
To gain a deeper understanding of the reaction path,the
following CASSCF(12,10)/MS-CASPT2 potential energy curves
(PECs) of the ground state and two low-lying excited states along
the representative trajectory are presented in Fig. 4. It is evident
that the two excited states S1 and S2 are almost degenerate at 0 fs,
corresponding to the dominate configurations HOMO -
- 1→LUMO and HOMO→LUMO,respectively. Corresponding
to the electron population in Fig. 3a,the electrons would be excited
to S1 from S0 at the beginning. At about 230 fs,the energy
difference between S1 and S0 is diminished to 0.503 eV (0.524 eV
for CASSCF),and the dominant configuration of S1 is changed to
HOMO→LUMO,consistent with the simulation findings in Fig. 3.
Then,these two states are obviously separate until they are
roughly degenerate again from 560 fs to 640 fs with the smallest
energy difference 0.576 eV (0.182 eV for CASSCF). Particularly,at
560 fs,the dominant configuration of S0 converts to HOMO -
→LUMO,while that for original S0 appears on S2 at CASSCF level,
although the dominant configurations are not changed for S0 and
S1 at MS-CASPT2 level. These results imply that electron transition
from S1 to S0 would occur at this moment,which is a little later
than 422 fs for the simulation in Fig. 3. Subsequently,these three
electronic states are not degenerate until the end.
In summary,we have investigated the photochromic ringopening
reaction mechanism of SP employing a realistic simulation
approach that couples the dynamics of electrons and nuclei and the
following SA3-CASSCF(12,10)/MS-CASPT2 PECs. The typical simulation
trajectory firstly involves two cooperative HOOP torsions of
two connected six membered rings. After the electronic transition
at about 560 fs,the C1-O bond is broken and the MC-TCC is formed
before 616 fs,at this moment the unstable product MC-CCC,
assigned to appear shortly after photo excitation,emerges and
rapidly converts back to the original MC product MC-TCC until
4848 fs. Then,the final stable TTC form ofMCis created and kept up
to the end. This means that the ring-opening process takes about
616 fs,which basically agrees with the 900 fs measured by the
experiments on the gas phase [9]. The subsequent isomerization on
the ground state takes about 4 ps,which is less than the about
20 ps obtained by the previous studies [10, 13]. This simulation
trajectory also provides a direct proof for confirming the proposed
mechanism suggested by Liu and Morokuma in more accurate
theoretical computations [19].
Acknowledgments
This work is supported by the National Natural Science
Foundation of China (Nos. 21003100 and 21073242),Natural
Science Basic Research Plan in Shaanxi Province of China (No.
2011JQ2013) and Special Fund of Education Department of
Shaanxi Province (No. 12JK0619). The authors also thank Professor
Zhenyi Wen,Associate Professor Huixian Han,and Bingbing Suo for
the helpful suggestions.

Scheme 1.The scheme of photo induced reaction between spiropyran and merocyanine.

Fig. 1. Snapshots taken at different times in a simulation of trans-SP in response to excitation by a 80 fs (FWHM) laser pulse with a fluence of 0.139 kJ/m2 and photon energy of
3.972 eV,Atom C,H,N,and O are respectively denoted by dark gray,French gray,blue,and red,(a)-(f) are at 0 fs,244 fs,422 fs,616 fs,4848 fs and 15,000 fs,respectively.

Fig. 2. Changes in (a) C1-O (b) the a,b and g (c) t and u of trans-SP subjected excitation by a 80 fs (FWHM) laser pulse with a fluence of 0.139 kJ/m2 and photon energy of
3.972 eV.

Fig. 3. Variations with time of (a) the electronic populations and (b) energies of theHOMO + 1,HOMO,LUMO and LUMO + 1 orbitals of trans-SP following application of a 80 fs
(FWHM) laser pulse with a fluence of 0.139 kJ/m2 and photon energy of 3.972 eV.

Fig. 4. Variations with time of potential energy curves (PECs) of the ground state and
two low-lying excited states along the representative trajectory at the level of
CASSCF(12,10)/MS-CASPT2 with the basis set 6-31G(d).
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| [1] | B.L. Feringa, Molecular Switches, Wiley-VCH, Weinheim, Germany, 2011, p. 476. |
| [2] | M. Heilemann, P. Dedecker, J. Hofkens, M. Sauer, Photoswitches: key molecules for subdiffraction-resolution fluorescence imaging and molecular quantification, Laser Photonics Rev. 3 (2009) 180-202. |
| [3] | J. Buback, M. Kullmann, F. Langhojer, et al., Ultrafast bidirectional photoswitching of a spiropyran, J. Am. Chem. Soc. 132 (2010) 16510-16519. |
| [4] | G. Berkovic, V. Krongauz, V. Weiss, Spiropyrans and spirooxazines for memories and switches, Chem. Rev. 100 (2000) 1741-1754. |
| [5] | S. Silvi, A. Arduini, A. Pochini, et al., A simple molecular machine operated by photoinduced proton transfer, J. Am. Chem. Soc. 129 (2007) 13378-13379. |
| [6] | (a) D.S. Achilleos, T.A. Hatton, M. Vamvakaki, Light-regulated supramolecular engineering of polymeric nanocapsules, J. Am. Chem. Soc. 134 (2012) 5726-5729; (b) Z. Si, Q. Zhang, M. Xue, et al., Synthesis of novel chalcone derivatives and their stabilization effect of spiropyran in PMMA films, Chin. Chem. Lett. 22 (2011) 1025-1028. |
| [7] | N.P. Ernsting, T. Arthen-Engeland, Conformational dynamics of the merocyanine, J. Phys. Chem. 95 (1991) 5502-5509. |
| [8] | N. Tamai, H. Miyasaka, Ultrafast dynamics of photochromic systems, Chem. Rev. 100 (2000) 1875-1890. |
| [9] | M. Rini, A.K. Holm, E.T.J. Nibbering, H. Fidder, Ultrafast UV-mid-IR investigation of the ring opening reaction of a photochromic spiropyran, J. Am. Chem. Soc. 125 (2003) 3028-3034. |
| [10] | L. Poisson, K.D. Raffael, B. Soep, J.M. Mestdagh, G. Buntinx, Gas-phase dynamics of spiropyran and spirooxazine molecules, J. Am. Chem. Soc. 128 (2006) 3169-3178. |
| [11] | M. Kullmann, S. Ruetzel, J. Buback, P. Nuernberger, T. Brixner, Reaction dynamics of a molecular switch unveiled by coherent two-dimensional electronic spectroscopy, J. Am. Chem. Soc. 133 (2011) 13074-13080. |
| [12] | J. Kohl-Landgraf, M. Braun, C.Özçoban, D.P.N. Gonçalves, A. Heckel, J. Wachtveitl, Ultrafast dynamics of a spiropyran in water, J. Am. Chem. Soc. 134 (2012) 14070- 14077. |
| [13] | Y. Sheng, J. Leszczynski, A.A. Garcia, et al., Comprehensive theoretical study of the conversion reactions of spiropyrans: substituent and solvent effects, J. Phys. Chem. B 108 (2004) 16233-16243. |
| [14] | K.P. Lawley, Advances in Chemical Physics, vol. 69, John Wiley & Sons Inc., New York, 1987, pp. 399-445. |
| [15] | J. Finley, P.A. Malmqvist, O. Roos, L. Serrano-andres, The multi-state CASPT2 method, Chem. Phys. Lett. 288 (1998) 299-306. |
| [16] | (a) Y. Kurashige, T. Yanai, High-performance ab initio density matrix renormalization group method: Applicability to large-scale multireference problems for metal compounds, J. Chem. Phys. 130 (2009) 234114; (b) D. Ghosh, J. Hachmann, T. Yanai, G.K.L. Chan, Orbital optimization in the density matrix renormalization group, with applications to polyenes and betacarotene, J. Chem. Phys. 128 (2008) 144117; (c) Y. Kurashige, T. Yanai, Second-order perturbation theory with a DMRG selfconsistent field reference function: theory and application to the study of chromium dimer, J. Chem. Phys. 135 (2011) 094104. |
| [17] | (a) P. Celani, F. Bernardi, M. Olivucci, M.A. Robb, Conical intersection mechanism for photochemical ring opening in benzospiropyran compounds, J. Am. Chem. Soc. 119 (1997) 10815-10820; (b) I. Gómez, M. Reguero, M.A. Robb, Efficient photochemical merocyanine-tospiropyran ring closure mechanism through an extended conical intersection seam. A model CASSCF/CASPT2 study, J. Phys. Chem. A 110 (2006) 3986- 3991. |
| [18] | M. Sanchez-Lozano, C.M. Estévez, J. Hermida-Ramón, L. Serrano-Andres, Ultrafast ring-opening/closing and deactivation channels for a model spiropyran-merocyanine system, J. Phys. Chem. A 115 (2011) 9128-9138. |
| [19] | (a) F.Y. Liu, K. Morokuma, Multiple pathways for the primary step of the spiropyran photochromic reaction: a CASPT2//CASSCF study, J. Am. Chem. Soc. 135 (2013) 10693-10702; (b) F.Y. Liu, Y. Kurashige, T. Yanai, K. Morokuma, Multireference Ab initio density matrix renormalization group (DMRG)-CASSCF and DMRG-CASPT2 study on the photochromic ring opening of spiropyran, J. Chem. Theory Comput. 9 (2013) 4462-4469. |
| [20] | V.I. Minkin, A.V. Metelitsa, I.V. Dorogan, et al., Spectroscopic and theoretical evidence for the elusive intermediate of the photoinitiated and thermal rearrangements of photochromic spiropyrans, J. Phys. Chem. A 109 (2005) 9605-9616. |
| [21] | (a) T. Chu, Y. Zhang, K. Han, The time-dependent quantum wave packet approach to the electronically nonadiabatic processes in chemical reactions, Int. Rev. Phys. Chem. 25 (2006) 201-235; (b) K. Han, J.D. Huang, S. Chai, S.H. Wen, W.Q. Deng, Anisotropic mobilities in organic semiconductors, Nat. Protoc. Exch. (2013), http://dx.doi.org/10.1038/ protex.2013.070. |
| [22] | Y.S. Dou, B.R. Torralva, R.E. Allen, Semiclassical electron-radiation-ion dynamics (SERID) and cis-trans photoisomerization of butadiene, J. Mod. Opt. 50 (2003) 2615-2643. |
| [23] | Y.S. Dou, B.R. Torralva, R.E. Allen, Another important coordinate in the photoisomerization of cis-stilbene, Chem. Phys. Lett. 378 (2003) 323-329. |
| [24] | T.B. Boykin, R.C. Bowen, G. Klimeck, Electromagnetic coupling and gauge invariance in the empirical tight-binding method, Phys. Rev. B 63 (2001) 245314- 245330. |
| [25] | B.R. Torralva, T.A. Niehaus, M. Elstner, et al., Response of C60 and Cn to ultrashort laser pulses, Phys. Rev. B 64 (2001) 153105-153108. |
| [26] | D. Porezag, T. Frauenheim, T. Köhler, D. Seifert, R. Kaschner, Construction of tightbinding- like potentials on the basis of density-functional theory: application to carbon, Phys. Rev. B 51 (1995) 12947-12957. |
| [27] | W.C. Swope, H.C. Andersen, P.H. Berens, K.R. Wilson, A computer simulation method for the calculation of equilibrium constants for the formation of physical clusters of molecules: applications to small water clusters, J. Chem. Phys. 76 (1982) 637-649. |
| [28] | G. Karlström, R. Lindh, P.Å. Malmqvist, et al., MOLCAS: a program package for computational chemistry, Comput. Matl. Sci. 28 (2003) 222-239. |