




b The CAS Key Laboratory of Receptor Research, Shanghai Institute of Materia Medica, Chinese Academy of Sciences, Shanghai 201203, China
1. Introduction
Glucocorticoids (GCs) exert a wide spectrum of metabolic and immunological effects via their cognate receptors (glucocorticoid receptors,GRs) which regulate a variety of important target genes [1]. Hypercortisolemia seen in Cushing’s disease or long-term therapy with GCs leads to a number of disease states,such as impaired glucose tolerance,hypertension,depression and central obesity [2]. Therefore,selective GR antagonists are useful in mitigating such conditions.
A well-known GR antagonist,mifepristone (RU486),binds to GR,PR (progesterone receptor) and AR (androgen receptor) with high affinities [3]. Treatment of diabetic mice with RU486 lowered the hepatic glucose level by reducing the expression of two key enzymes,phosphoenolpyruvate carboxykinase (PEPCK) and glucose 6-phosphatase (G6Pase) involved in gluconeogenesis [4, 5]. Data from Abbott Laboratories demonstrated that liver-selective GR antagonist (e.g.,the cholate conjugate of RU486) could significantly decrease blood glucose levels in ob/ob mice indicating that GR modulation may have value in the control of metabolic disorders [6, 7].
We have previously reported the discovery and characterization of a small,non-steroidal molecule GR antagonist 1a (Fig. 1, IC50 = 165.7 nmol/L) identified during a high-throughput screening (HTS) campaign [8]. Compound 1a had a 20-fold selectivity on hGR (human glucocorticoid receptor) over hPR (human progesterone receptor),nearly 28-fold selectivity on hGR over hMR (human mineralocorticoid receptor),and no activity on hAR (human androgen receptor) and hER (human estrogen receptor). As a follow-up study,we describe here the hit-to-lead optimization of 1a and its allyl derivative 8d,as a novel GR antagonist.

|
Download:
|
| Fig. 1.Structure of 1a. | |
2. Experimental
Chemistry: Reagents were commercial grade and were used as received,unless otherwise noted. NMR spectra were recorded on Varian Mercury 300 or Varian Inova 600 spectrometers. Chemical shifts were reported in parts per million (ppm),with the solvent resonance as the internal standard (CDCl3 7.26 ppm,CD3OD 3.31 ppm,DMSO-d6 2.50 ppm for 1H NMR; DMSO-d6 39.52 ppm for 13C NMR). Silica gel (200-300 mesh,Shanghai Chemical Reagents Co.,Ltd.) was used for column chromatography. Silica gel 60 F254 plates (Qingdao Ocean Chemical Industry Co.,Ltd.) were used for TLC analysis. Ozone generator BGF-YQ (Beijing Guoke Ozone Application Technology Co.,Ltd.) was used for ozonization.
Bioassay: Dexamethasone (Dex) and RU486 were purchased from Sigma-Aldrich Chemical Co. (St. Louis,MO,USA) and tritiated Dexamethasone ([3H]Dex) from GE Healthcare Life Sciences (Chalfont St. Giles,Bucks.,UK). Fetal bovine serum (FBS) and charcoal/dextran-treated FBS (CDT-FBS) were bought from Hyclone (Logan,UT,USA). Steady-Glo Luciferase Assay System was obtained from Promega Corporation (Madison,WI,USA) and AlamarBlueTM from US Biological (Salem,MA,USA). FlexStation 384II was the product of Molecular Devices (Sunnyvale,CA,USA).
Experimental procedures and 1H NMR and 13C NMR of all the compounds can be found in Supporting information.
3. Results and discussion Our initial strategy was to investigate the impact of substitutions on the phenyl ring of the 1,2,3,4,4a,9a-hexahydro-1Hxanthene skeleton. The key intermediates 4a-j,prepared from bimorpholine aminals of the substituted salicylicaldehydes, underwent [4 + 2] cycloaddition with 1-morpholinocyclohexene to yield the titled compounds 1a-j according to the procedures described by Ukhin et al. (Scheme 1) [9, 10]. Compound 1a was then hydrogenated to give 1k,which was then acetylated to afford the amide analog 1l. Next,we evaluated the influence of the active hydrogen on the 2-methylindole group. Compounds 5a-c were synthesized by direct alkylation of 1a or 1b with propyl bromide (5a),benzyl bromide (5b) and methyl iodide (5c),respectively. Efforts were also made to replace 2-methylindole with other groups,such as phenylethynyl,substituted phenyl,quinolin-4-yl, or pyrrol-3-yl [11, 12],which resulted in the total loss of activity (data not shown). Then,modification of cyclohexane (6a-h) was implemented by a similar method (Scheme 2),in which cyclohexanyl was converted into phenyl[1, 2]cyclohexanyl (6a), 2-methylcyclohexanyl (6b),cyclopentanyl (6c and 6d) or cycloheptanyl (6e-h) using the corresponding enamines. Finally,effects of the 4a-morpholinyl group was examined by the introduction of other N-containing heterocyclic groups,such as piperidinyl (6i), piperazinyl (6j),pyrrolidinyl (6k),or alkylated amino groups,such as N,N-dipropylamino (6l) (Scheme 2). Hydrolyzation of 4amorpholinyl led to hemiketal 7a,which was subsequently refluxed with pyridinium 4-toluenesulfonate (PPTS) in the mixture of benzene and methanol (2:1,v/v) to obtain the 4a-OMe substituted compound 8a. Other substitutions,such as hydrogen (8b-c),allyl (8d and 8e) and CN (8g),were introduced by the reaction of hemiketal 7a or 7b with substituted silico reagents (e.g., triethylsilane [13],trimethylallylsilane [14] or trimethylcyanosilane [15]). Hydrogenation of 8e afforded the 4a-propyl derivative 8f. After that,compound 8d was used as the intermediate for the subsequent modification by ozonization [16] to obtain the acetaldehyde 9,which allowed a further SAR exploration of 10a-d by the Wittig reaction. Reduction of 9 with sodium borohydride afforded the alcohol 11a,which was followed by acetylation in acetic anhydride to yield 11b. Iodization of 11a followed by elimination in DBU gave the 4a-vinyl substituted compound 13 (Scheme 3).

|
Download:
|
| Scheme 1.The synthetic routes for compounds 1a-j and 5a-c. Reagents and conditions: (i) morpholine,i-PrOH,reflux,77%; (ii) 5-substituted 2-methylindole,130 ℃ 59%; (iii) 4-(cyclohex-1-en-1-yl) morpholine,165 ℃,17-77%; (iv) COONH4,MeOH,Pd/C,r.t.,74%; (v) Ac2O,H2O r.t.,96%; (vi) PrBr (5a) or BnBr (5b) or MeI (5c),NaH,THF,r.t.,20-47%. | |

|
Download:
|
| Scheme 2.The synthetic routes for compounds 6a-l. Reagents and conditions: (i) substituted enamine,165 ℃,12-74%. | |

|
Download:
|
| Scheme 3.The synthetic routes for compounds 8a-g,9,10a-e,11a-b,12 and 13. Reagents and conditions: (i) AcOH,H2O,reflux,70-74%; (ii) 7a,MeOH,PPTS,benzene/ methanol (2:1),reflux,48%; (iii) 7a or 7b,Et3SiH,BF3-Et2O,DCM,-20 ℃,then aq NaHCO3,85-89%; (iv) 7a or 7b,trimethylallylsilane,TiCl4,DCM,-78 ℃,then aq NH4Cl,23-25%; (v) 7a,TMSCN,TMSOTf,-78 ℃ to reflux,then aq NaHCO3,5%; (vi) H2,10% Pd/C,THF,64%; (vii) (1) Boc2O,4-DMAP,DCM; (2) O3,-78 ℃,then Ph3P,-78 ℃ to r.t.; (3) TFA,DCM,0 ℃ to r.t.,35% for 3 steps in total; (viii) (1) EtBr (10a) or BuBr (10b) or iBuBr (10c),PPh3; (2) BuLi,THF,-78 ℃ to 0 ℃; (3) -78 ℃,THF,then aq NH4Cl,26-35%; (ix) ethyl 2-(triphenylphosphoranylidene)propanoate,toluene,reflux,71%; (x) NaOH,THF/MeOH/H2O (4:2:1),reflux,61%; (xi) NaBH4,THF/MeOH (10:1),r.t.,85%; (xii) Ac2O,H2O,r.t.;(xiii) Ph3P,1-H-imidazole,I2,toluene,reflux,83%; (xiv) DBU,benzene,reflux,61%. | |
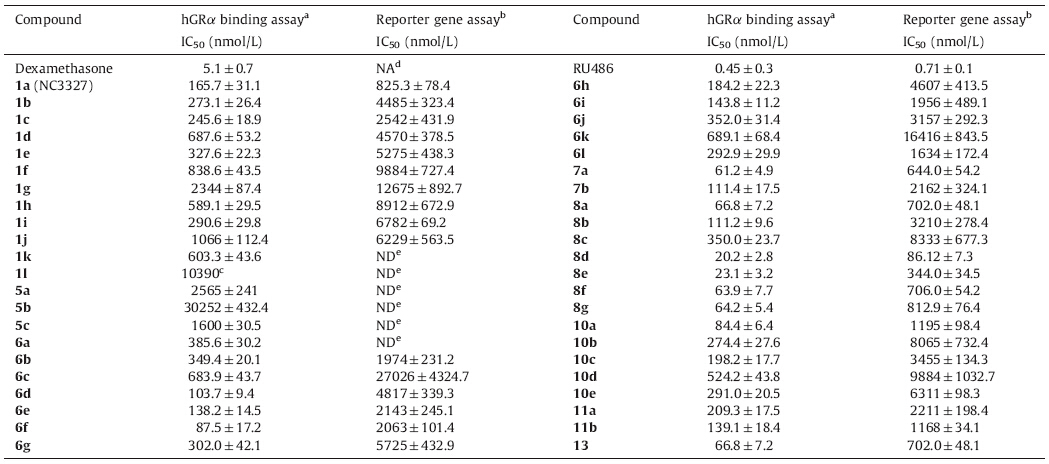
All these compounds were examined for their binding affinities to hGRa using [3H]Dex as the probe and their effects on the transcriptional activity mediated by GR were assessed with a luciferase reporter gene co-transfection assay in CV-1 cells. The results showed that modification of the phenyl ring of 1,2,3,4,4a,9a-hexahydro-1H-xanthene with 7-nitro improved the binding affinity and cellular activity by 2-fold and 5-fold, respectively (1a vs. 1b). Other electron-withdrawing groups,such as chloro or bromo substitution,had limited impacts on the assay (1c and 1e vs. 1b) with the 7-chloro derivative (1c) exhibiting nearly equal binding affinity to GR and 40% improved cellular activity compared with the 7-unsubstituted compound (1b). On the contrary,7-substituents of electron-donating groups displayed an opposite picture. Activities of compounds substituted by 7-OCF3 (1g),7-OCH3 (1h) or 7-NH2 (1k) lost their activities significantly in both assays,especially 7-OCF3 (1g),whose IC50 was only 2344 × 87.4 nmol/L,indicating that electron-donating groups with modest steric hinderance at this position were unfavorable for the receptor binding. Substitution on position 5 did not render any advantages for both GR binding and reporter gene activities, regardless of electron-withdrawing (1d and 1f),or donating groups (1i). As for modification of the indole group,it was determined that alkylation of the nitrogen caused a dramatic decrease in potency (5a and 5b vs. 1b and 5c vs. 1a),and substitution of 50-methoxy on the 50- position markedly reduced the potency by 5-fold (1j vs. 1c) (Table 1). The influence of the size of the cycloalkane was also evaluated by changing from cyclopentyl to cycloheptyl (6d,6e vs. 1c). Compared with cyclohexyl (1c),substitutions of cyclopentyl (6d) and cycloheptyl (6e) improved the binding affinity by nearly 50%,while the cellular activity was either decreased (6d vs. 1c),or remained unchanged (6e vs. 1c). Similar results were also observed in 7-nitro substituted compounds (6f vs. 1a). Finally,the influence of 4amorpholine group was assessed by replacing it with several other groups. Compounds 6i-k were synthesized by replacement of 4amorpholine with 1-piperidinyl (6i),1-piperazinyl (6j) or 1-pyrrolidinyl (6k),respectively. Binding assays illustrated a profile indicating substitutions of saturated heterocycles were more potent than aromatic ones,which is consistent with the results in the reporter gene assay (6i-k vs. 1c). It appears that the 4a-morpholine group was not an essential group,as its removal did not alter bioactivity (8b vs. 1a and ℃ vs. 1c). Enhanced activities were recorded in compounds with 4a-hydroxyl (7a vs. 1a and 7b vs. 1c),or 4a-methoxy (8a vs. 1a) suggesting that reduced hindrance may be responsible for this observation. Notably,substitution of 4a-allyl resulted in an 8.2-fold increase in binding potency accompanied by a 9.6-fold better cellular activity (8d vs. 1a),whereas shortening of the chain length led to a reduction of binding by 3-fold and of luciferase activity by 8-fold, respectively (13 vs. 8d). The same phenomenon was seen in compounds 8e and 1c. Hydrogenation of the allyl group resulted in decreases of GR binding and cellular activities (8f vs. 8e). Terminal alkylation of the allyl group caused a significant reduction of bioactivities in both assays (10a-c vs. 8d) especially for the propyl (10b) or isopropyl (10c) substitution,indicating that introduction of sterically large groups at the terminal olefinic link was inappropriate for binding to GR.
| Table 1 Results of binding and reporter gene assays for the analogs 1,5-8,10-11 and 13. |
{kind=link}
{kind=link}
{kind=link}
{kind=link}
4. Conclusion
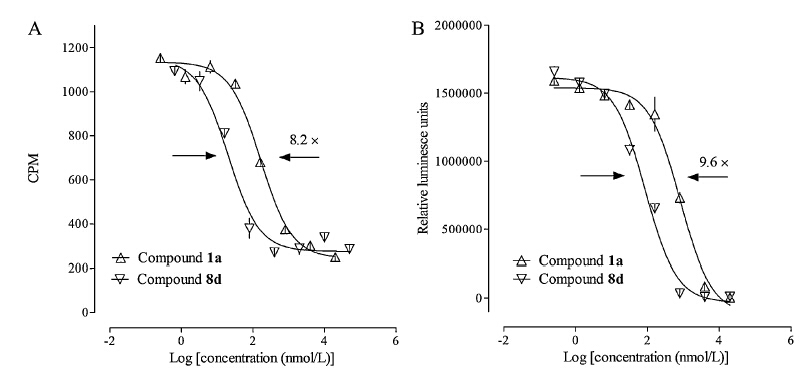
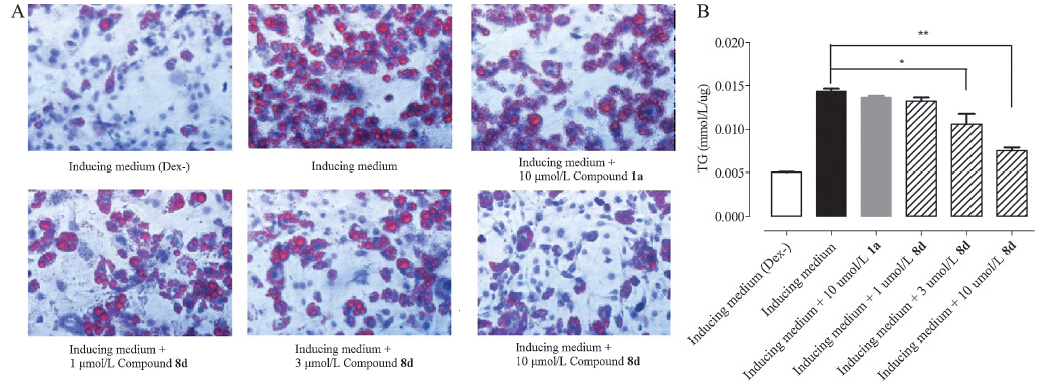
Optimization of the initial hit compound 1a guided by the SAR analysis identified an improved GR antagonist 8d (Fig. 2A) that did not possess any agonist profile in the cell-based reporter gene assay (data not shown). In the antagonist mode,both compounds 1a and 8d suppressed dexamethasone-mediated cellular activities with 8d displaying a higher potency (Fig. 2B). Like 1a,compound 8d was capable of (1) reducing dexamethasone-induced transcription of two key enzymes in gluconeogenesis,PEPCK and G6Pase,in HepG2 cells (Fig. 3) and (2) reducing dexamethasone-induced 3T3-L1 preadipocyte differentiation (Fig. 4),both are indicative of GR antagonism. Although 8e was less active in cell-based reporter gene assay,itmight represent a more promising lead due to the existence of the nitro group on 8d. Furthermore,4a-alkyl substitution led to an improved chemical stability comparedwith the original structure of the O,N-ketal,which could easily be hydrolyzed in acid solution under high temperature. Improved antagonistic activity and chemical stability make this scaffold a promising starting point for subsequent optimization,including further studies on 4asubstitution and effects of specific enantiomers.

|
Download:
|
| Fig. 2. (A) Glucocorticoid receptor binding characteristics of 1a and 8d. (B) Glucocorticoid receptor mediated transcriptional activities of 1a and 8d. | |
{kind=link}

|
Download:
|
| Fig. 3. Effects of 1a and 8d on glucocorticoid receptor (GR)-mediated gluconeogenesis. HepG2 cells were grown in 6-well plates and incubated with various compounds for 24 h. Total RNA was isolated with TRIzol reagent. (A) Phosphoenolpyruvate carboxykinase (PEPCK) and (B) glucose-6-phosphatase (G6Pase) mRNA levels in HepG2 cells were quantified by real-time RT-PCR. GAPDH was used as an internal control. Dexamethasone (Dex) at 0.01 mmol/L could increase the expression of PEPCK and G6Pase,both were rate-controlling enzymes of gluconeogenesis. As antagonists of human GRa,1a and 8d were able to reduce Dex-induced transcription,and the effect of 3 mmol/L 8d was statistically better than that of 1a at the same concentration. ##P < 0.01 compared with 0.1 mmol/L Dex-treated cells; *P < 0.05 compared with 0.1 mmol/L Dex plus 3 mmol/L 1a. Data (means ± SEM) are representative of three independent experiments. | |
{kind=link}

|
Download:
|
| Fig. 4. Effects of 1a and 8d on differentiation of 3T3-L1 preadipocytes. 3T3-L1 cells were grown in 6-well plates and allowed to reach confluence. Various concentrations of 1a and 8d were added into the medium and incubated for 1 h. Differentiation was induced by changing medium to the inducing medium containing 10% FBS,0.5 mmol/L IBMX, 1 mg/mL insulin,0.3 mmol/L dexamethasone (Dex) and human GRa antagonists (1a or 8d). After two days,the inducing medium were changed to insulin medium and incubated for two more days. At last,cells were cultured in DMEM containing 10% FBS for four days. Following differentiation,(A) cells were stained with Oil Red O and representative images were obtained at 200× original magnification,and (B) triglyceride produced by 3T3-L1 cells was measured,which was normalized by protein content in the cells. *P < 0.05 and **P < 0.01 compared with cells cultured in inducing medium. Data (means ± SEM) are representative of three independent experiments. | |
{kind=link}
Acknowledgments
We are indebted to Dale E. Mais for critical review of this manuscript. The study was supported in part by grants from the Ministry of Health of China (Nos. 2012ZX09304-011, 2013ZX09401003-005,2013ZX09507001 and 2013ZX09507002), Shanghai Science and Technology Development Fund (No. 13DZ2290300) and Thousand Talents Program in China.
Appendix A. Supplementary data
Supplementary data associated with this article can be found,in the online version,at http://dx.doi.org/10.1016/j.cclet.2014.03.017.
| [1] | R.D. Clark, Glucocorticoid receptor antagonists, Curr. Top. Med. Chem. 8 (2008) 813-838. |
| [2] | A. McMaster, D.W. Ray, Drug insight: selective agonists and antagonists of the glucocorticoid receptor, Nat. Clin. Pract. Endocrinol. Metab. 4 (2008) 91-101. |
| [3] | L.G. Hamann, L.J. Farmer, M.G. Johnson, et al., Synthesis and biological activity of novel nonsteroidal progesterone receptor antagonists based on cyclocymopol monomethyl ether, J. Med. Chem. 39 (1996) 1778-1789. |
| [4] | T. Gettys, P.Watson, I. Taylor, S. Collins, RU-486(Mifepristone)ameliorates diabetes but does not correct deficient beta-adrenergic signalling in adipocytes from mature C57BL/6J-ob/ob mice, Int. J. Obes. Relat. Metab. Disord. 21 (1997) 865-873. |
| [5] | J.E. Friedman, Y. Sun, T. Ishizuka, et al., Phosphoenolpyruvate carboxykinase (GTP) gene transcription and hyperglycemia are regulated by glucocorticoids in genetically obese db/db transgenic mice, J. Biol. Chem. 272 (1997) 31475-31481. |
| [6] | T.W. von Geldern, N. Tu, P.R. Kym, et al., Liver-selective glucocorticoid antagonists: a novel treatment for type 2 diabetes, J. Med. Chem. 47 (2004) 4213-4230. |
| [7] | J. Link, B. Sorensen, J. Patel, et al., Antidiabetic activity of passive nonsteroidal glucocorticoid receptor modulators, J. Med. Chem. 48 (2005) 5295-5304. |
| [8] | Q.Y. Li, M. Zhang, T.M. Hallis, et al., Characterization of a novel non-steroidal glucocorticoid receptor antagonist, Biochem. Biophys. Res. Commun. 391 (2010) 1531-1536. |
| [9] | L.Y. Ukhin, L. Belousova, V. Khrustalev, Reactions of 2-methylindole with morpholinals of substituted salicylaldehydes, Russ. Chem. Bull. 52 (2003) 700-704. |
| [10] | L.Y. Ukhin, L. Belousova, Z.I. Orlova, S. Shishkina, O. Shishkin, Reactions of nitrogenous derivatives of substituted salicylaldehydes with cyclic ketones and enamines, Russ. Chem. Bull. 51 (2002) 1262-1269. |
| [11] | L.Y. Ukhin, V. Komissarov, S. Lindeman, V. Khrustalev, Y.T. Struchkov, Aminals in the synthesis of 1 3-substituted propargylamines and 3H-2-vinylidene-3-aminobenzofuran derivatives, Russ. Chem. Bull. 43 (1994) 413-419. |
| [12] | L. Jurd, Synthesis of new benzopyrans related to podophyllotoxin, J. Heterocycl. Chem. 26 (1989) 1349-1352. |
| [13] | F.A. Carey, H.S. Tremper, Carbonium ion-silane hydride transfer reactions. I. Scope and stereochemistry, J. Am. Chem. Soc. 90 (1968) 2578-2583. |
| [14] | J.W. Kennedy, D.G. Hall, Recent advances in the activation of boron and silicon reagents for stereocontrolled allylation reactions, Angew. Chem. Int. Ed. Engl. 42 (2003) 4732-4739. |
| [15] | C. Bolm, P. Müller, Enantioselective trimethylsilylcyanation of aldehydes, Tetrahedron Lett. 36 (1995) 1625-1628. |
| [16] | R.K. Boeckman Jr., M.D. Shair, J.R. Vargas, L.A. Stolz, Synthetic and mechanistic studies of the retro-Claisen rearrangement. 2. A facile route to medium-ring heterocycles via rearrangement of vinylcyclopropane and cyclobutanecarboxaldehydes, J. Org. Chem. 58 (1993) 1295-1297. |