




Aromatic halides are an important class of chemicals which has been extensively used in modern organic syntheses and pharma- ceutical industry [1]. Currently the most prevalent approaches to access aromatic halides still highly rely on the classic protocols [2], such as eletrophilic aromatic substitution,directed ortho-lithiation and Sandmeyer reaction,etc. However,those methodologies always suffer from one or more limitations including harsh reaction conditions,poor regioselectivity,moisture sensitivity,low yield,and tedious reaction operations,etc. In recent years, transition metals catalyzed C-H functionalization reactions through weak coordination had emerged as powerful tools for aromatic carbon-halide bond formation [3]. For example,this strategy has been successfully applied to prepare aromatic iodides with substrates,such as phenyl acetic acids,benzoic acids,and sulfonamides by palladium catalysis.
More recently,we developed a palladium(II)-catalyzed ortho- halogenation reaction to prepare a variety of arene halides with electron-deficient arenes and NBS/NCS [4]. In our studies,ester groups were found to work as an effective directing group for promotingorthoC-Hactivationandthisprotocolcanbe conducted in mild conditions without the need for moisture- or air-proof conditions. The efficiency and utility of this method has also been demonstrated in an iterative ortho-halogenation reaction and large-scale synthesis of some simple aryl halides. Here,we report a synthetic study of the gram-scale synthesis of multi-substituted aryl halides through both palladium catalyzed C-H halogenation reactions and traditional methods respectively. The aryl halides can be further transformed into (6-amino-2-chloro-3-fluorophe- nyl)methanol,which is a useful synthetic intermediate employed in medicinal chemistry and organic electronic material. For example,this compound will serve as a basic building block for the OLED (organic light-emitting diode) material synthesis. A brief comparison between these two strategies was summarized to highlight the advantages of our new C-H halogenation method.
2. Experimental 2.1. Synthesis of compounds 2-5 via C-H activation approachEthyl 2-bromo-5-fluorobenzoate (2): Compound 1 (1.68 g, 1.0 equiv.), NBS (1.87 g, 1.05 equiv.), Pd(OAc)2 (78.5 mg, 0.035 equiv.) and Na2S2O8(2.86 g,1.2 equiv.) were dissolved in commercial dichloroethane (12 mL) in a 100 mL round bottom flask. Then TfOH (2.2 mL,2.5 equiv.) was added into the reaction solution. Then the reaction mixture was stirred at 80 ℃ for 9 h. After completion of the reaction,the mixture was cooled to room temperature and then saturated NaHCO3was added to quench the reaction. The reaction mixture was diluted with dichloromethane and washed once with saturated aqueous NaHCO3. Then organic layer was dried over Na2SO4and concentrated on rotavapor under reduced pressure. Finally the residue was purified by silical gel column chromatography to give compound 2 (1.71 g) in 70% yield.
Ethyl 6-bromo-2-chloro-3-fluorobenzoate (3): Compound 3 was prepared with similar procedure as compound 2. Compound 2 (1.45 g,1.0 equiv.),NCS (0.83 g,1.05 equiv.),Pd(OAc)2(52.9 mg, 0.04 equiv.),Na2S2O8(1.68 g,1.2 equiv.),TfOH (2.6 mL,5.0 equiv.) wereused.Thereactionmixturewasstirredat80 ℃for4 h.Finally, compound 3 (0.78 g) was obtained in 47% yield.
Ethyl 6-amino-2-chloro-3-fluorobenzoate (4): Compound 3 (560 mg,1.0 equiv.),Cu2O (28.6 mg,0.1 equiv.),ammonium hydroxide (25%) (3.1 mL,10.0 equiv.) were dissolved in NMP (5 mL) in a sealed tube. The reaction mixture was stirred at 80 ℃ for 12 h. With similar workup as above,finally,compound 4 (390 mg) was obtained in 90% yield.
(6-Amino-2-chloro-3-fluorophenyl)methanol (5): LiAlH4 (50 mg,2.2 equiv.) in anhydrous THF (1.32 mL) was added dropwise to compound 4 (130 mg,1.0 equiv.) in anhydrous THF (4 mL) at 0 ℃. Then the mixture was added at room temperature for 6 h. Finally compound 5 (63 mg) was obtained in 60% yield.
2.2. Synthesis of compounds of 7-5 via traditional approach2-Chloro-3-fluorobenzoic acid (7): A solution of 3-fluoroben- zoic acid (6,4.02 g,1.0 equiv.) in 20 mL of THF was added dropwise to a suspension of TMEDA (10.0 mL,2.3 equiv.) and 1.5 mol/L n- BuLi (42.1 mL,2.2 equiv.) in 50 mL of THF at -78 ℃. The mixture was stirred at -78 ℃ for 30 min,and then a solution of hexachloroethane (2.72 g,4.0 equiv.) in 50 mL of THF was added. After 12 h,the reaction was quenched with water and diluted with diethyl ether. The bilayer was adjusted to pH 1-2 with conc. hydrochloric acid. The organic layer was washed with water,brine, dried and concentrated to give crude as a tan solid,which was washed with hexane to give of the desired product 7 (3.0 g) in 60% yield.
2-Chloro-3-fluoro-6-nitrobenzoic acid ( 8 ): In a three necked flask fitted with a dropping funnel and a thermometer were charged compound 7 (5 g,1.0 equiv.) and concentrated H2SO4 (43 mL). After cooling to 0 ℃,HNO3 (70%,3.3 mL) was added dropwise over 30 min,keeping the temperature between 0 to 10 ℃. After 1 h,the reaction mixture was poured into the crushed ice keeping the temperature below 20 ℃. The mixture was extracted with EtOAc,the combined extract was washed with brine and dried over Na2SO4. The filtrate was evaporated to give mixture (5.35 g) of 8 and 80(ratio 2:1) and the total yield was 85%. The mixture was used to synthesize compound 9 directly without further purification.
(2-Chloro-3-fluoro-6-nitrophenyl)methanol ( 9 ): Compound 8 and 80(2 g,1.0 equiv.) were dissolved in toluene (10 mL). SOCl2 (1.99 mL,3.0 equiv.)wasthenaddedandthemixturewasstirredat reflux for 3 h under argon. The solvent was evaporated and the crude chloride was directly reduced. A suspension of NaBH4 (346 mg,1.0 equiv.) in THF/DMF (15 mL/15 mL,1:1) under argon was cooled to 0 ℃,then the solution of crude chloride in dry THF was added dropwise. Themixture was stirred atroom temperature for 24 h. The reaction was quenched by slowly addition of 10% HCl and extracted with Et2O. The combined organic layer were washed with brine,dried over Na2SO4and concentrated. The mixture was purified by a short column chromatography to give a pure mixture (1.25 g,ratio 2:1) of compound 9 and 90. The yield for compound 9 determined by1H NMR was 40% (749 mg).
(6-Amino-2-chloro-3-fluorophenyl)methanol (5): Compound 9 and 90(1.60 g,1.0 equiv.),iron powder (2.40 g,5.5 equiv.) and NH4Cl (0.30 g,0.7 equiv.) were suspended in ethanol/water (10:1) and refluxed for 2 h. The solvent was evaporated and DCM was added. The mixture was washed with a saturated solution of sodium carbonate,dried over Na2SO4,filtered and concentrated in vacuo. Finally the crude was purified by silical gel column chromatography to give the compound 5 (458 mg) in a yield of 76%.
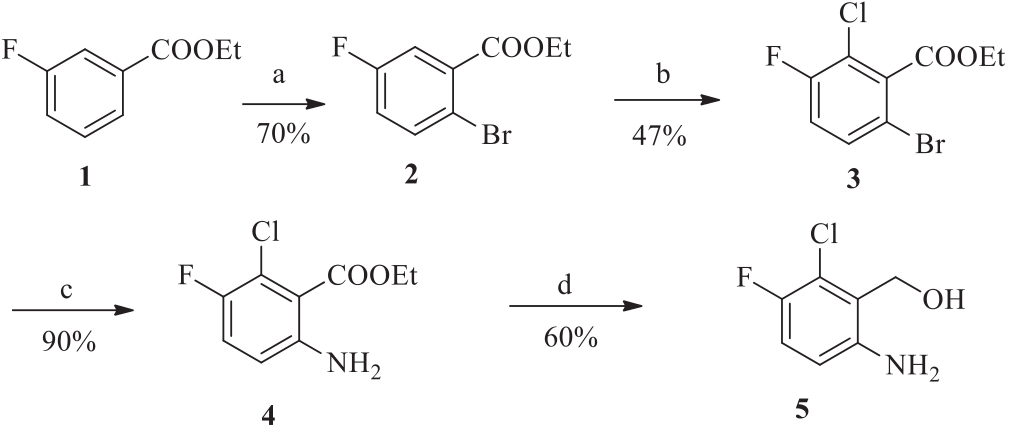
3. Results and discussionAt the beginning of our synthetic studies towards (6-amino-2- chloro-3-fluorophenyl)methanol,we envisioned two halide atoms can be introduced into both ortho-positions (2- and 6-) of 3- fluorobenzoates through weak coordination assisted C-H haloge- nation. A following aminolysis manipulation can selectively transform the 6-halide group into an amino functional group. As depicted in Scheme 1,we started our investigation with our newly developed iterative C-H halogenation methods. Starting with a simple ethyl 3-fluorobenzoate,brominated product 2 can be smoothly produced via the first C-H bromination on a gram scale in 70% yield. Then,a second C-H chlorination reaction was conducted under the optimum reaction conditions. We were pleased to find that dihalogenated intermediate 3 can be successfully obtained in a yield of 47% on a gram scale with an excellent regioselectivity. It is noteworthy to point out that the second chlorination reaction is quite challenging because of the intrinsic electron-poor property and high steric hindrance of compound 2 from the first bromination step. The amount of TfOH was a key factor for C-H halogenations. 2.5 equiv. of TfOH was sufficient to promote the reaction in the bromination step. However,more TfOH (5.0 equiv.) was needed in the second chlorination step,which might be due to the electron-deficiency of arene2.Withthecompound3inhand,weconductedaminolysisto selectively replace bromine atom into amino group,which can readily give intermediate 4 in an excellent yield (90%) [5]. A reduction with lithium aluminium hydride can finally furnish desired product 5 in a good yield of 60% [6]. The overall yield was 18% in four steps. As shown in Scheme 2,a synthetic comparison study of compound 5 with classical methods was also conducted in parallel. With 3-fluorobenzoic acid 6 as the starting material,this approach involved ortho-lithiation [7],nitration [8] and a sequential reduction [9, 10],which can provide product 5 in a total yield of 10% (4 steps).

|
Download:
|
| Scheme 1. C-H activation strategy for the synthesis of (6-amino-2-chloro-3- fluorophenyl)methanol. (a) Pd(OAc)2,NBS,TfOH,Na2S2O8,DCE,80 ℃; (b) Pd(OAc)2, NCS,TfOH,Na2S2O8,DCE,80 ℃; (c) NH4OH,Cu2O,NMP/H2O,80 ℃; (d) LiAlH4,THF. | |
{kind=link}

|
Download:
|
| Scheme 2. Traditional approach for the synthesis of (6-amino-2-chloro-3- fluorophenyl)methanol. (a) n-BuLi,TMEDA,C2Cl6,THF,-78 ℃; (b) conc. HNO3, conc. H2SO4; (c) i. SOCl2,toluene,reflux; ii. NaBH4,THF/DMF,r.t.; (d) Fe,NH4Cl, EtOH/H2O (10:1),reflux. | |
{kind=link}
In comparison to the synthetic strategy employing C-H halogenation,based on our experimental results,a few limitations from traditional methods were summarized as followings: (1) It was not easy to do the first chlorination reaction with ortho- lithiation method because of the requirement of absolute anhydrous conditions and low temperature (-78 ℃) for a long period of time. Furthermore,safety issue is another concern since a large amount of strong base (n-BuLi) and toxic hexochloroethane were used in the reaction. (2) The nitration step using conc. H2SO4 and conc. HNO3suffered from poor selectivity and gave a mixture of two regioisomers,which caused a serious purification problem. (3) The workup of nitro group reduction step was not trivial. As iron power was used as the reductant,a long time was consumed to do the filtration.
4. ConclusionIn summary,a practical and efficient approach has been developed to prepare (6-amino-2-chloro-3-fluorophenyl)metha- nol through iterative C-H halogenation reactions. Compared with traditional methods,the new strategy employing C-H activation methods demonstrated a few advantages such as milder reaction conditions,higher yields,better selectivity and practicality,and high chemical diversity,etc. As the field of transition metal catalyzed C-H functionalization keeps moving forward,we expect more efficient and practical reactions could be developed in the near future and this promising strategy will play a more important role in organic synthesis.
AcknowledgmentsThis work was supported by ‘973’ Project (No. 2011CB965300), NSFC(Nos.21142008,21302106),TsinghuaUniversity985PhaseII Funds and the Tsinghua University Initiative Scientific Research Program. We thank Prof. L. Liu (Tsinghua University) for helpful discussions.
| [1] | (a) N. Sotomayor, E. Lete, Aryl and heteroaryllithium compounds by metalhalogen exchange. Synthesis of carbocyclic and heterocyclic system, Curr. Org. Chem. 7 (2003) 275-278; (b) A. Butler, J.V. Walker, Marine haloperoxdases, Chem. Rev. 93 (1993) 1937- 1944; (c) S.V. Ley, A.W. Tomas, Modern synthetic methods for copper-mediated C (aryl)- O, C (aryl)-N, and C (aryl)-S bond formation, Angew. Chem. Int. Ed. 42 (2003) 5400- 5449; (d) K.C. Nicolaou, P.G. Bulger, D. Sarlah, Palladium-catalyzed cross-coupling reactions in total synthesis, Angew. Chem. Int. Ed. 44 (2005) 4442-4489; (e) K.C. Nicolaou, P.G. Bulger, D. Sarlah, Palladiumkatalysierte kreuzkupplungen in der total synthese, Angew. Chem. 117 (2005) 4516-4563; (f) J. Hassen,M. Sevignon, C. Gozzi, E. Schulz, M. Lemaire, Aryl-aryl bond formation one century after the discovery of Ullmann reaction, Chem. Rev. 102 (2002) 1359- 1470; (g) Q.L. Shen, T. Ogata, J.F. Hartwig, Highly reactive, general and long-lived catalysts for palladium-catalyzed amination of heteroaryl and aryl chlorides, bromides, and iodides: scope and structure-activity relationships, J. Am. Chem. Soc. 130 (2008) 8564; (h) M. Gorjizadeh, S. Sayyahi, A novel and efficient synthesis of alkyl thiocyanates from alkyl halides in water using phase transfer catalysts, Chin. Chem. Lett. 22 (2011) 659-662; (i) X.Wu, W. Hu, Direct synthesis of diaryl sulfides by copper-catalyzed coupling of aryl halide with aminothiourea, Chin. Chem. Lett. 23 (2012) 391-394. |
| [2] | (a) S. Ma, G. Villa, P.S. Thuy-Boun, A. Homa, J.Q. Yu, Pd-catalyzed ortho-selective C-H deuteration of arenes: evidence for superior reactivity of weakly coordinated palladacycles, Angew. Chem. Int. Ed. 53 (2014) 734-737; (b) K.M. Engle, T.S. Mei, M. Wasa, J.Q. Yu, Weak coordination as a powerful means for developing broadly useful C-H functionalization reactions, Acc. Chem. Res. 45 (2012) 788-802; (c) T.S. Mei, R. Giri, N. Maugel, J.Q. Yu, Pd-catalyzed mono-selective orthohalogenation of C-H bonds assisted by counter cations: an orthogonal method to directed ortho-lithiation, Angew. Chem. Int. Ed. 47 (2008) 5215-5219; (d) X.Y. Sun, Y.H. Sun, C. Zhang, Y. Rao, Room-temperature Pd-catalyzed C-H chlorination by weak coordination: one-pot synthesis of 2-chlorophenols with excellent regioselectivity, Chem. Commun. 50 (2014) 1262-1264; (e) G. Shan, X.L. Yang, L.L. Ma, Y. Rao, Pd-catalyzed C-H oxygenation with TFA/ TFAA: expedient access to oxygen-containing heterocycles and late-stage drug modification, Angew. Chem. Int. Ed. 51 (2012) 13070-13074. |
| [3] | (a) W.E. Parham, C.K. Bradsher, Aromatic organolithuim reagents bearing electrophilic groups. Preparation by halogen-lithuim exchange, Acc. Chem. Res. 15 (1982) 300-305; (b) J. Mortier, J. Moyroud, B. Bennetau, P.A. Cain, The carboxylic acid group as an effective director of ortho-lithiation, J. Org. Chem. 59 (1994) 4042-4044. |
| [4] | X.Y. Sun, G. Shan, Y.H. Sun, Y. Rao, Regio- and chemoselective C-H chlorination/ bromination of electron deficient arenes by weak coordination and study of relative directing-group abilities, Angew. Chem. Int. Ed. 52 (2013) 4440- 4444. |
| [5] | H.H. Xu, C. Wolf, Efficient copper-catalyzed coupling of aryl chlorides, bromides and iodides with aqueous ammonia, Chem. Commun. 21 (2009) 3035-3037. |
| [6] | M.Q. Jia, S.L. You, N-Heterocyclic carbene-catalyzed enantioselective intramolecular N-tethered aldehyde-ketone benzoin reactions, Catalysis 3 (2013) 622-624. |
| [7] | J. Busch-Petersen, T.B. Leonard, M.R. Palovich, et al., Treatment of cystic fibrosis or the symptoms associated with cystic fibrosis, U.S. Pat. Appl. Publ. 20100255167. |
| [8] | C. Tachdjian, D.S. Karanewsky, X.Q. Tang, et al., Modulation of chemosensory receptors and heterobicyclic ligands associated therewith and their preparation, U.S. Pat. Appl. Publ. 20110224155. |
| [9] | A. Blanc, C.G. Bochet, Isotopic effects in photochemistry: application to chromatic orthogonality, Org. Lett. 9 (2007) 2649-2651. |
| [10] | M.P. Storz, C.K. Maurer, C. Zimmer, et al., Validation of PqsD as an anti-biofilm target in Pseudomonas aeruginosa by development of small-molecule inhibitors, J. Am. Chem. Soc. 134 (2012) 16143-16146. |