




b Unit of Science and Technology, Faculty of Science, King Khalid University, Abha 61413, Saudi Arabia;
c Center of Excellence for Advanced Materials Research, King Khalid University, Abha 61413, Saudi Arabia;
d Chemistry Department, Faculty of Science, King Abdulaziz University, Jeddah 21589, Saudi Arabia;
e Center of Excellence for Advanced Materials Research, King Abdulaziz University, Jeddah 21589, Saudi Arabia
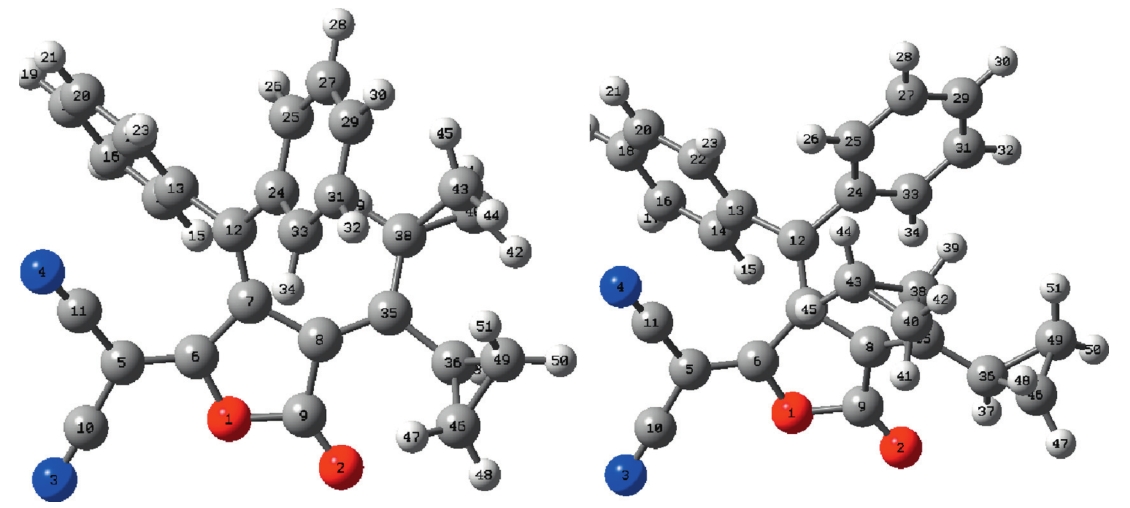
Previously [1] we shed light on the color change behavior of the title compound by tribochromism (color change due to mechanical friction). Now we introduce the crystallochromism (color change due to the change of crystal habit) and solvation chromism. Our recent quantum chemical study revealed that crystallochromism and solvation chromism would also be the major factors due to which the title compound,3-dicyclopropylmethylene-5-dicyano- methylene-4-diphenylmethylenetetra-hydrofuran-2-one, gave yellow (1Y) and red (1R) crystals (Figs. 1 and S1,and Table S1 in Supporting information),while both give red solutions. Previously,we showed that in 1Y the diphenylmethylene groups are folded,i.e.,one phenyl lies above a cyclopropyl group and the other above a cyano group,while in 1R the diphenylmethylene groups have a twisted orientation (one phenyl lies above a cyclopropyl group and the other below a cyano group) [1]. Moreover,we showed that the important calculated red and blue shifts correspond to the cases where the Al-N bonds are affected in thesame direction,i.e.,lengtheningorshorteningat thesametime. The lengthening of Al-N bonds causes a red shift while shortening results in a blue shift [2].

|
Download:
|
| Fig. 1. Optimized structures of 1R (left) and 1Y (right) at B3LYP/6-31G** level of theory. | |
The ground state geometries were optimized at DFT/B3LYP/6- 31G** [3] level of theory with Gaussian09 package [4]. The frequencycalculationswere carriedout at the same levelof theory. The absorption spectra were computed by using TD-DFT/B3LYP/6- 31G** level of theory.
3. Results and discussionSignificant differences in bond length (C7-C12),bond angles (C6-C5-C11 and C35-C38-C40) and dihedral angles have been observed between 1R and 1Y,see Table S1 (details can be found in Supportinginformation,labelingFig.S2).Ingeneral,C5-C6,C8-C35 and C7-C12 bond lengths being lengthened in 1R,i.e.,0.016,0.013 and 0.036 A˚,respectively,while O1-C9,C35-C38 and C12-C13 are being lengthened in 1Y,i.e.,0.013,0.021,and 0.023 A˚,respectively. The following bond angles being increases in 1R; C6-C5-C11,C35- C38-C40 and C35-C38-C43,i.e.,2.66,2.39 and 1.81 A˚,respectively, while C7-C12-C13 (1.79 A˚) in 1Y. The noteworthy difference has been observed in the dihedral angles,e.g. in1Y C1-C6-C5-C11,C8- C35-C36-C46,C7-C12-C13-C14 are 58,1138,208 more twisted than the 1R,respectively. The C1-C6-C5-C10 and C8-C35-C38- C43 of 1R are being 38,1018 more twisted than 1R. In 1R,C7-C8- C35-C36 and C7-C12-C24-C25 are 1738 and 1468 while in 1Y ?1688 and ?1188. In 1R,C8-C35-C38-C40,C8-C35-C36-C49 and C7-C12-C24-C33are?1528,?1118and?388whilein1Y1068,1388 and 668,respectively. In our previous study [2],we pointed out that theimportantcalculatedredandblueshiftscorrespondtothecases where the Al-N bonds are affected in the same direction,i.e., lengtheningorshorteningatthesametime.ThelengtheningofAl-N bonds cause red shift while shortening result blue shift. In 1R, prominent lengthening has been observed 0.036 A˚. Moreover,we noticedthatthebondanglesgenerallyincreasesinthe1R.Similarly dihedral angle is also playing its role by twisting. As a whole it is expected that lengthening of bond lengths and superior the bond anglesin1Rthanthatof1Yarealsothecausesthat1Risredone.The yellow compound has the dipole moment 9.47 D while red one 11.66 D. Beck [5] and coworkers described that di[1,1-bis(dicya- nomethylene)-indan-2-ylidene] can exist in yellow and in red crystal variations. Theyexplained that inyellowcrystals,molecules have a folded structure,while red molecules have a twisted structure. The delocalization of the highest occupied molecular orbital (HOMO) in 1R is higher than the 1Y. The charge distribution on HOMO and lowest unoccupied molecular orbital (LUMO) on the right phenyl ringishigherin1Rthan 1Y(Fig. 2).The1Y absorb light in the region 363 nm with the transition H to L,while 1R absorbs at 345 (H-3 to L) and 528 nm (H to L) (Fig. S3 in Supporting information). It showed that 1R is absorbing light also at longer wavelengths. The main vibration in IR has been observed at 1592 cm?1for mode429,while 1Y at 1635 cm?1for mode 219 (Fig. S4 in Supporting information). It is suspected that the red of 1R mightbeduetothelargebonddistances,superiorbondangles,more charge distributionon frontier molecular orbitals and absorption in longer wavelength compared to 1Y.

|
Download:
|
| Fig. 2.Frontier molecular orbitals of 1Y (top) and 1R (bottom). | |
In the next phase,we tried to explain why,in solution,both are red or in other words,yellow turns to red solution. We optimized 1Yintolueneandfoundnosignificantdifferenceinthegeometrical parameters between solvent optimized geometry and optimized geometry without solvent,see Table 1. Moreover,we computed the absorption wavelength in toluene for 1Y and found that the absorption wavelengths of 1Y with,and without,solvent are identical. Fig. 3 illustrates that in 1Y close contacts through CN have been observed (CN to CN),while in 1R no near contacts with other functional groups were found. It is expected that the intermolecular interactions between the CN parts of the 1Y crystal which are overlapping,but these are very weak and in solution again become red due to the absence of intermolecular interaction. Finally,1Y turningto a red solution might be due to tribochromism and solvation chromism. Due to the crystallochromism 1Y and 1R are yellow and red,respectively,because the crystal structures are entirely different.
| Table 1 The bond lengths (A˚),bond angles and dihedral angles (degree) for 1Y at ground state (S0) at B3LYP/6-31G** level of theory. |
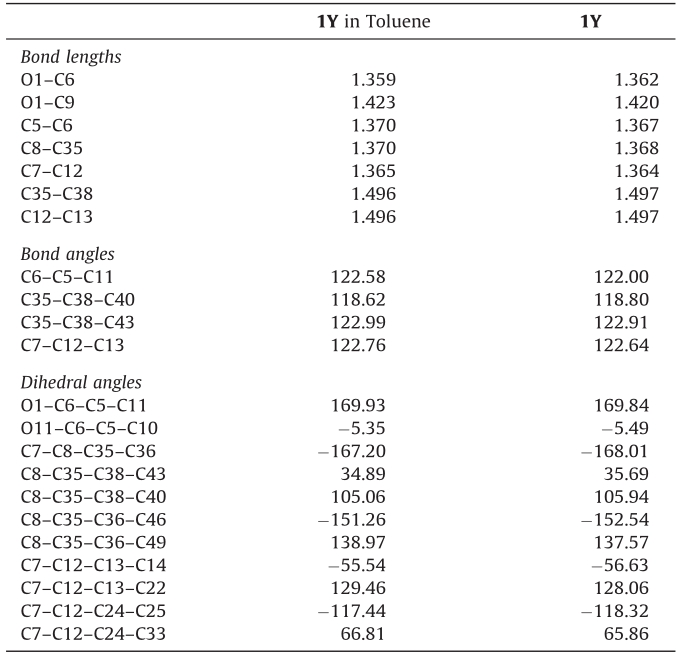
{kind=link}
{kind=link}

|
Download:
|
| Fig. 3. The near contact representation of CN-CN in 1Y (left) and 1R (right). | |
{kind=link}
The crystals of 1Y and 1R have different bond lengths,bond angles,dihedral angles,charge distribution on highest occupied molecularorbital,lowestunoccupiedmolecularorbitals,absorption andIRspectra.Due tothese entirefactors,one showedyellowwhile the second exhibited red crystals. There exist very weak intermo- lecularinteractionsbetweentheCNpartsofthe1Ycrystal,butbythe mechanical crushing 1Y again becomes red with the absence of intermolecular interactions. The solvation chromism is also the causethat1Yturnstoredsolution.The1Yand1Rareyellowandred in crystals,respectively,due to the crystallochromism.
AcknowledgmentsThe support and facilities to carry out the research work provided by King Khalid University are greatly acknowledged.
Appendix A. Supplementary dataSupplementary data associated with this article can be found,in the online version,at http://dx.doi.org/10.1016/j.cclet.2014.01.016.
| [1] | A.M.A. Asiri, H.G. Heller, M.B. Hursthouse, A. Karalulov, Tribochromic compounds, exemplified by 3-dicyclopropylmethylene-5-dicyanomethylene-4-diphenylmethylenetetrahydrofuran- 2-one, Chem. Commun. (2000) 799-800. |
| [2] | A. Irfan, R. Cui, J. Zhang, L. Hao, Push-pull effect on the charge transfer, and tuning of emitting color for disubstituted derivatives of mer-Alq3, Chem. Phys. 364 (2009) 39-45. |
| [3] | (a) A.G.Al-Sehemi, A. Irfan,A.M.Asiri,TheDFTinvestigations of the electroninjection in hydrazone-based sensitizers, Theor. Chem. Acc. 131 (2012) 1199-1208; (b) A.G. Al-Sehemi, M.A.M. Al-Melfi, A. Irfan, Electronic, optical, and charge transfer properties of donor-bridge-acceptor hydrazone sensitizers, Struct. Chem. 24 (2013) 499-506; (c) A. Irfan, A.G. Al-Sehemi, Quantum chemical study in the direction to design efficient donor-bridge-acceptor triphenylamine sensitizers with improved electron injection, J. Mol. Model. 18 (2012) 4893-4900; (d) R. Jin, A. Irfan, Theoretical study of coumarin derivatives as chemosensors for fluoride anion, Comput. Theor. Chem. 986 (2012) 93-98; (e) A. Irfan, N. Hina, A.G. Al-Sehemi, A.M. Asiri, Quantum chemical investigations aimed at modeling highly efficient zinc porphyrin dye sensitized solar cells, J. Mol. Model. 18 (2012) 4199-4207; (f) A.G. Al-Sehemi, A. Irfan, A.M. El-Agrody, Synthesis, characterization and DFT study of 4H-benzo[h]chromene derivatives, J. Mol. Struct. 1018 (2012) 171-175; (g) A. Irfan, F. Ijaz, A.G. Al-Sehemi, A.M. Asiri, Quantum chemical approach toward rational designing of highly efficient oxadiazole based oligomers used in organic field effect transistors, J. Comput. Electron 11 (2012) 374-384; (h) A.D. Becke,Density-functional thermochemistry. ⅡI.The role of exact exchange, J. Chem. Phys. 98 (1993) 5648-5652; (i) B. Miehlich, A. Savin, H. Stoll, H. Preuss, Results obtained with the correlation energy density functionals of Becke and Lee, Yang and Parr, Chem. Phys. Lett. 157 (1989) 200-206; (j) C. Lee, W. Yang, R.G. Parr, Development of the Colle-Salvetti correlationenergy formula into a functional of the electron density, Phys. Rev. B 37 (1988) 785-789. |
| [4] | M.J. Frisch, G.W. Trucks, H.B. Schlegel, et al., Gaussian 09, Revision A.1, Gaussian, Inc., Wallingford, CT, 2009. |
| [5] | A. Beck, R. Gompper, K. Polborn, H.U. Wagner, Bi[1,3-bis(dicyanomethylene)indan- 2-ylidene]-an ethylene derivative with extremely pronounced, twisting of the CC Bond, Angew. Chem. Int. Ed. Engl. 32 (1993) 1352-1354. |