




1. Introduction
Recently,the introduction of chirality into tetrathiafulvalene (TTF) received much attention and stimulated arise and rapid development of new research into so-called chiral conductors and multifunctional materials. Various synthetic strategies for introduction of chirality into TTF have been proposed [1],and a large number of chiral TTF donors have been synthesized [2]. While the majority of synthesized chiral TTF derivatives have been utilized in preparation of conducting charge-transfer (CT) complexes and radical cation salts [3],they are also interesting as redox-active ligands for asymmetric synthesis [4],as chiroptical donors [5],and in formation of organogel and supramolecular chirality [6]. We have designed a novel silyl-substituted chiral TTF donor based on the following conception. Introduction of chalcogens in a peripheral position is one of the most effective strategies for chemical modification of TTF,to increase the dimensionality and consequently improve the conductivity of the CT salts. In this respect incorporation of silyl groups to TTF is an alternative way, though less exploited compared to thiolate or thioether,as reported by Avarvari et al. [7]. The new TTF derivative containing a disiloxane motif may also be used in multifunctional materials combining two or more physical properties,such as conductivity and molecular sensing (towards metal ions),considering the possible complexation effect of the S,O,Si-heterocycle in the structure. Herein,we report the synthesis and electrochemical properties of the new silyl-substituted chiral TTF derivative.
2. Experimental
All chemical reagents were purchased from commercial sources and used as received unless otherwise stated. Melting points were determined on a WRS-2A capillary melting apparatus,and the quoted temperatures were uncorrected. 1H NMR and 13C NMR spectra were recorded on a Bruker AM 400 spectrometer. CDCl3 was used as the solvent and chemical shifts recorded were internally referenced to Me4Si (0 ppm). IR spectra were obtained on a Thermo Electron Corporation Nicolet 380 FT-IR spectrophotometer. UV-vis spectra were recorded on a Hitachi U-3310 spectrophotometer in CH2Cl2 (c = 10-5 mol L-1) at room temperature. Mass spectra were recorded on a JEOL JMS-SX102A spectrometer using electron ionization (EI) at 70 eV or an Agilent 6000 LC-MS instrument (ESI,positive mode,70-1000 amu). Optical rotations were recorded at 589 nm on a Rudolph Autopol I polarimeter using a 1 dm cell,and [a]D values are given in 10-1 deg cm2 g-1. X-ray crystal structure was measured on a Bruker Smart CCD diffractometer by using Mo Ka radiation at 296(2) K. Voltammetric analysis was carried out in a standard three-electrode cell with a CHI760 potentiostat at 293 K. The working electrode was a platinum disc electrode and the auxiliary electrode was a platinum wire. The reference electrode was a saturated calomel electrode (SCE,0.1 mol L-1 Bu4NClO4 in CH2Cl2). The scanning speed was 100 mV s-1. Spectroscopic data of new compounds 7-9 were as follows:
7: Yellow crystals,mp 76-78 ℃; 1H NMR (400 MHz,CDCl3): d 0.20 (s,12H,CH3),2.06 (s,4H,CH2); 13C NMR (100 MHz,CDCl3): d 2.2 (SiCH3),24.2 (SiCH2),143.2 (C55C),213.8 (C55S); MS (EI,70 eV): m/z (%) 356 (M+,90),149 (100),163 (50),131 (37),70 (31),164 (30),173 (30),134 (23); IR (KBr,cm-1): υ 2916,1255,1071,836.
8: Pale yellow solid,mp 85-86 ℃; 1H NMR (400 MHz,CDCl3): d 0.19 (s,12H,SiCH3),2.02 (s,4H,SiCH2); 13CNMR(100 MHz,CDCl3): d 0.9 (SiCH3),22.4 (CH2),132.7 (C=C),199.8 (C=O); MS (ESI): m/z 304.4 (M+ + 1); IR (KBr,cm-1): υ 2953,2896,1664,1376,1250, 1155,1083,1071,740 cm-1.
9: Orange solid,mp 191-193 ℃; 1H NMR (400 MHz,CDCl3): d 0.19 (s,12H,SiCH3),1.40 (s,6H,CH3),1.98 (s,4H,SiCH2),2.64 (dd, 2H,J = 13.1,12.4 Hz),3.26 (dd,2H,J = 13.1,3.8 Hz),4.22 (dd,2H, J = 12.4,3.8 Hz); 13C NMR (100 MHz,CDCl3): σ 3.26 (SiCH3),24 (SiCH2),29 (CH3),39 (CH2),82 (CH),112 (OCO),134 (C=C),135 (C=C); IR (KBr,cm-1): υ 2986,2953,2913,2892,1636,1411,1252, 1113,1209,1071,842,797,705; [a]20 D + 79.6 (c 0.02,CHCl3).
3. Results and discussion
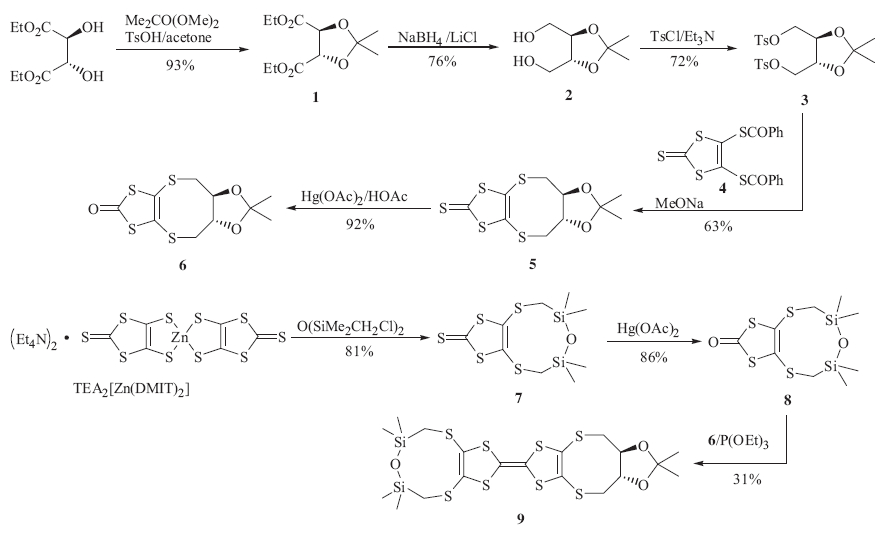
The synthetic route to chiral TTF derivative 9 was showed in Scheme 1. Preparation of compound 1 involves the protection of 1,2-diol. It was reported that compound 1 could be prepared by refluxing a solution of diethyl L-tartrate,p-toluenesulfonic acid and 2,2-dimethoxypropane in benzene [8]. We tried to use toluene instead of toxic benzene as solvent to synthesize 1 but found that more by-products were formed,decreasing yield of 1. Feit [9] prepared 1 from diethyl L-tartrate and acetonewhich plays a dual-effect of reagent and solvent. However,the reaction took nine days to complete.We improved synthesis of 1 by refluxing a solution of diethyl L-tartrate,p-toluenesulfonic acid and 2,2- dimethoxypropane in acetone. The reaction was finished in several hours to afford 1 in a high yield of 93%. Reduction of compound 1 with sodium borohydride and lithium chloride gave 2 in 76% yield. Sodium borohydride alone does not reduce esters under ambient conditions,but the reactivity of sodium borohydride can be enhanced by the addition of certain additives such as lithium chloride [10] and iodine [11]. Conversion of 2-3 was achieved by tosylation of 2 with p-toluene sulfonyl chloride in the presence of triethylamine [12]. Thione 5 was synthesized by referring to literature wherein ditosylate 3 was reacted with disodium salt of 1,3-dithiole-2-thione-4,5-dithiolate in THF for five days to afford 5 in 52% yield [13]. We tried to modify the procedure in order to synthesize 5 in a higher yield and shorter time. At first,wecarried out the reaction in methanol.A solution of sodium methoxide in methanol was added to compound 4 to generate disodium 1,3-dithiole-2-thione-4,5-dithiolate,to which 3 was added and the suspension was stirred for two days. In this case thione 5 was obtained in a low yield due to the poor solubility of 3 in methanol. Then we carried out the reaction in a mixed solvent of methanol/THF to get thione 5 in two days in 63% yield. The thionewas converted to the corresponding 1,3-dithiole-2-one 6 by reaction with mercuric acetate in high yield. Reaction between TEA2[Zn(DMIT)2] and 1,3-bis(chloromethyl)-1,1,3,3- tetramethyldisiloxane in acetone produced thione 7 in 81% yield then it was converted to the corresponding 1,3-dithiole- 2-one 8 by reaction with mercuric acetate in 86% yield. Triethyl phosphite-mediated cross-coupling between 6 and 8 afforded the new chiral TTF derivative 9 in 31% yield. The 1H NMR and 13C NMR spectrum (Fig. S1 and Fig. S2,respectively in Supporting information) of chiral TTF 9 were obtained and all the spectral data were in good accordance with the structure of the new compound.

|
Download:
|
| Scheme 1.Synthetic route to chiral TTF derivative 9.580 G.-Q. Liang et al. / Chinese Chemical Letters 25 (2014) 579-582 | |
{kind=link}
Single crystal structure of 8 is shown in Fig. 1. Compound 8 crystallizes in the triclinic system with space group P-1 (Table S1 in Supporting information). The C(1)-O(1) distance [1.205(2) A˚] is within the normal range of a typical C55O double bond (Table S2 in Supporting information). The C-S bond lengths out of the fivemembered dithiole ring are 1.825(3) and 1.815(2) A˚ for C(4)-S(3) and C(9)-S(4) bond,respectively. While the C-S bond lengths in the five-membered ring are shorter than a typical C-S single bond [range 1.746(2)-1.772(2) A˚]. The C=C bond length [1.343(3) A˚] in the five-membered ring is slightly longer than a normal double bond. This shows the high degree of conjugation of the fivemembered dithiole ring moiety in compound 8 (Fig. S3 in Supporting information).

|
Download:
|
| Fig. 1.Crystal structure of [(Fig._2)TD$FIG] compound 8. | |
{kind=link}
The electrochemical properties of the chiral TTF derivative 9 were studied in methylene dichloride solution by cyclic voltammetry and the cyclic voltammogram of 9 in methylene dichloride solution (10-3 mol L-1) was shown in Fig. 2. The new TTF derivative exhibited two reversible redox waves. The half-wave oxidation potentials of 9 (E1/21 and E1/22) are larger than those of TTF but are close to those of BEDT-TTF [14]. The difference between E1/21 and E1/22 (ΔE = E1/22 - E1/21) for 9 is smaller than those for TTF and BEDT-TTF,indicative of decreased Coulombic repulsion in the dicationic redox state of 9. The HOMO level of 9 was estimated from the first onset of the oxidation potential [EHOMO = -(Eox onset + 4.4) eV] [15] to be -5.11 eV,which makes it suitable for use as p-type organic semiconductor [16]. Therefore compound 9may be a good precursor for preparation of conducting CT complexes and radical cation salts.

|
Download:
|
| Fig. 2.Cyclic voltammogram of the chiral TTF derivative 9 in CH2Cl2. | |
{kind=link}
The UV-vis absorption spectra of donor 9 and 9-DDQ CT complex in methylene dichloride (10-5 mol L-1) were measured at room temperature (Fig. S4 in Supporting information). Donor 9 alone showed two strong absorption peaks at 325 nm and 405 nm. Upon mixing of 9 with DDQ,a new CT band at 425 nm was visible, confirming the formation of CT complex between 9 and DDQ. It demonstrated the usefulness of the new donor for preparation of CT complexes with suitable electron acceptors such as DDQ. Further studies in this aspect are underway. 4. Conclusion
In conclusion,a new silyl-substituted chiral TTF derivative and related 1,3-dithiole-2-(thi)one compounds were synthesized. Crystal structure of the silyl-substituted 1,3-dithiole-2-one revealed the high degree of conjugation of the five-membered ring in the compound. Cyclic voltammetry of the chiral TTF derivative showed that its electron-donating ability was similar to that of BEDT-TTF. The DE value for the new TTF derivative was smaller than those for TTF and BEDT-TTF,indicative of decreased Coulombic repulsion in the dicationic redox state. Formation of CT complex between the new donor and DDQ was demonstrated. Apart from as a promising precursor for conducting CT complexes and salts,the new TTF derivative may be used in multifunctional materials. Acknowledgments
The authors acknowledge the support by ‘‘the Fundamental Research Funds for the Central Universities’’.
Appendix A. Supplementary data
Supplementary data associated with this article can be found,in the online version,at http://dx.doi.org/10.1016/j.cclet.2014.01.018.
| [1] | N. Avarvari, J.D. Wallis, Strategies towards chiral molecular conductors, J. Mater. Chem. 19 (2009) 4061-4076. |
| [2] | (a) A. Saad, F. Barrière, E. Levillain, et al., Persistent mixed-valence [(TTF)2]+* dyad of a chiral bis(binaphthol) tetrathiafulvalene (TTF) derivative, Chem. Eur. J. 16 (2010) 8020-8028; (b) S.J. Yang, A.C. Brooks, L. Martin, et al., Novel enantiopure bis(pyrrolo)tetrathiafulvalene donors exhibiting chiral crystal packing arrangements, CrystEngComm 11 (2009) 993-996; (c) M. Chas, F. Riobé, R. Sancho, C. Minguillon, N. Avarvari, Selective monosulfoxidationof tetrathiafulvalenes intochiralTTF-sulfoxides,Chirality21(2009)818-825; (d) C. Réthoré, A. Madalan, M. Fourmigué, et al., O×S vs. N×S intramolecular nonbonded interactions in neutral and radical cation salts of TTF-oxazoline derivatives: synthesis, theoretical investigations, crystalline structures, and physical properties, New J. Chem. 31 (2007) 1468-1483; (e) C. Réthoré, M. Fourmigué, N. Avarvari, Tetrathiafulvalene-hydroxyamides and - oxazolines: hydrogen bonding, chirality, and a radical cation salt, Tetrahedron 61 (2005) 10935-10942; (f) C. Réthoré, M. Fourmigué, N. Avarvari, Tetrathiafulvalene based phosphinooxazolines: a new family of redox active chiral ligands, Chem. Commun. (2004) 1384-1385; (g) S. Matsumiya, A. Izuoka, T. Sugawara, T. Taruishi, Y. Kawada, Effect of methyl substitution on conformation andmolecular arrangement of BEDT-TTF derivatives in the crystalline environment, Bull. Chem. Soc. Jpn. 66 (1993) 513-522; (h) J.D. Wallis, A. Karrer, J.D. Dunitz, Chiral metals? A chiral substrate for organic conductors and superconductors, Helv. Chim. Acta 69 (1986) 69-70. |
| [3] | (a) F. Pop, P. Auban-Senzier, A. Fraąckwiak, et al., Chirality driven metallic versus semiconducting behavior in a complete series of radical cation salts based on dimethyl-ethylenedithio-tetrathiafulvalene (DM-EDT-TTF), J. Am. Chem. Soc. 135 (2013) 17176-17186; (b) F. Pop, S. Laroussi, T. Cauchy, et al., Tetramethyl-bis(ethylenedithio)-tetrathiafulvalene (TM-BEDT-TTF) revisited: crystal structures, chiroptical properties, theoretical calculations, and a complete series of conducting radical cation salts, Chirality 25 (2013) 466-474; (c) A. Saad, O. Jeannin, M. Fourmigué, Chiral, flexible binaphthol-substituted tetrathiafulvalenes, Tetrahedron 67 (2011) 3820-3829; (d) A.M. Madalan, C. Réthoré,M. Fourmigué, et al., Order versus disorder in chiral tetrathiafulvalene-oxazoline radical-cation salts: structural and theoretical investigations and physical properties, Chem. Eur. J. 16 (2010) 528-537; (e) M. Chas, M. Lemarié, M. Gulea, N. Avarvari, Chemo- and enantioselective sulfoxidation of bis(ethylenedithio)-tetrathiafulvalene (BEDT-TTF) into chiral BEDT-TTF-sulfoxide, Chem. Commun. (2008) 220-222; (f) C. Réthoré, N. Avarvari, E. Canadell, P. Auban-Senzier, M. Fourmigué, Chiral molecular metals: syntheses, structures, and properties of the AsF6 - salts of racemic (±)-, (R)-, (S)-tetrathiafulvalene-oxazoline derivatives, J. Am. Chem. Soc. 127 (2005) 5748-5749; (g) A. Karrer, J.D. Wallis, J.D. Dunitz, et al., Structures and electrical properties of some new organic conductors derived fromthe donormolecule TMET (S,S,S,Sbis( dimethylethylenedithio)tetrathiafulvalene), Helv. Chim. Acta 70 (1987) 942-953. |
| [4] | (a) C. Réthoré, F. Riobé, M. Fourmigué, et al., Tetrathiafulvalene-oxazoline ligands in the iridium catalyzed enantioselective hydrogenation of arylimines, Tetrahedron: Asymmetry 18 (2007) 1877-1882; (b) C. Réthoré, I. Suisse, F. Agbossou-Niedercorn, et al., Chiral tetrathiafulvalene based phosphine- and thiomethyl-oxazoline ligands. Evaluation in palladium catalysed asymmetric allylic alkylation, Tetrahedron 62 (2006) 11942-11947; (c) A. Chesney, M.R. Bryce, Chiral oxazolines linked to tetrathiafulvalene (TTF): redox-active ligands for asymmetric synthesis, Tetrahedron: Asymmetry 7 (1996) 3247-3254. |
| [5] | (a) T. Biet, A. Fihey, T. Cauchy, et al., Ethylenedithio-tetrathiafulvalene-helicenes: electroactive helical precursors with switchable chiroptical properties, Chem. Eur. J. 19 (2013) 13160-13167; (b) Y.L. Si, G.C. Yang, Z.M. Su, Chiroptical, linear, and second-order nonlinear optical properties of tetrathiafulvalenylallene: a multifunctional molecular material, J. Mater. Chem. C 1 (2013) 1399-1406; (c) M. Hasegawa, Y. Sone, S. Iwata, H. Matsuzawa, Y. Mazaki, Tetrathiafulvalenylallene: a new class of donor molecules having strong chiroptical properties in neutral and doped states, Org. Lett. 13 (2011) 4688-4691. |
| [6] | (a) C. Wang, F. Sun, D.Q. Zhang, et al., Cholesterol-substituted tetrathiafulvalene (TTF) compound: formation of organogel and supramolecular chirality, Chin. J. Chem. 28 (2010) 622-626; (b) I. Danila, F. Riobé, J. Puigmartí-Luis, et al., Supramolecular electroactive organogel and conducting nanofibers with C3-symmetrical architectures, J. Mater. Chem. 19 (2009) 4495-4504; (c) I. Danila, F. Pop, C. Escudero, et al., Twists and turns in the hierarchical self-assembly pathways of a non-amphiphilic chiral supramolecular material, Chem. Commun. 48 (2012) 4552-4554; (d) I. Danila, F. Riobé, F. Piron, et al., Hierarchical chiral expression from the nanoto mesoscale in synthetic supramolecular helical fibers of a nonamphiphilic C3- symmetrical p-functional molecule, J. Am. Chem. Soc. 133 (2011) 8344-8353. |
| [7] | (a) A. Hameau, F. Guyon, M. Knorr, et al., Synthesis and reactivity of silylated tetrathiafulvalenes, Dalton Trans. 36 (2008) 4866-4876; (b) F. Biaso, M. Geoffroy, E. Canadell, et al., Intramolecular mixed-valence state through silicon or germanium double bridges in rigid bis(tetrathiafulvalenes), Chem. Eur. J. 13 (2007) 5394-5400. |
| [8] | (a) M. Carmack, C.J. Kelley, The synthesis of the optically active Cleland reagent[(±)-1,4-dithio-Lg-threitol], J. Org. Chem. 33 (1968) 2171-2173; (b) A.F. Rodney, An efficient synthesis of (±)-posticlure: the sex pheromone of Orgyia postica, Eur. J. Org. Chem. (2007) 5064-5070; (c) P.M. Chincholkar, A.S. Kale, V.K. Gumaste, A.R.A.S. Deshmukh, An efficient formal synthesis of (S)-dapoxetine from enantiopure 3-hydroxy azetidin-2-one, Tetrahedron 65 (2009) 2605-2609. |
| [9] | P.W. Feit, 1,4-Bismethanesulfonates of the stereoisomeric butanetetraols and related compounds, J. Med. Chem. 7 (1964) 14-17. |
| [10] | A.E. Wróblewski, I.E. Góowacka, Enantiomerically pure 4-amino-1,2,3-trihydroxybutylphosphonic acids, Tetrahedron 61 (2005) 11930-11938. |
| [11] | M. Periasamy, M. Thirumalaikumar, Methods of enhancement of reactivity and selectivity of sodium borohydride for applications in organic synthesis, J. Organomet. Chem. 609 (2000) 137-151. |
| [12] | G. Liu, Z. Wang, Total synthesis of koninginin D, B and E, Synthesis (2001) 119-127. |
| [13] | G.A. Horley, T. Ozturk, F. Turksoy, J.D. Wallis, New substrates for the preparation of electroactive materials: the syntheses of chiral tetrathiafulvalene derivatives with hydroxyfunctionalised butane-1,4-dithio bridges, J. Chem. Soc., Perkin Trans. 1 (1998) 3225-3231. |
| [14] | K. Zong, W. Chen, M.P. Cava, R.D. Rogers, Synthesis and properties of bis(2,5- dimethylpyrrolo[3,4-d]) tetrathiafulvalenes, a class of annelated tetrathiafulvalene derivatives with excellent electron donor properties, J. Org. Chem. 61 (1996) 8117-8124. |
| [15] | L. Wang, H. Cho, S.H. Lee, et al., Liquid crystalline mesophases based on symmetric tetrathiafulvalene derivatives, J. Mater. Chem. 21 (2011) 60-64. |
| [16] | A.R. Murphy, J.M.J. Fréchet, Organic semiconducting oligomers for use in thin film transistors, Chem. Rev. 107 (2007) 1066-1096. |