




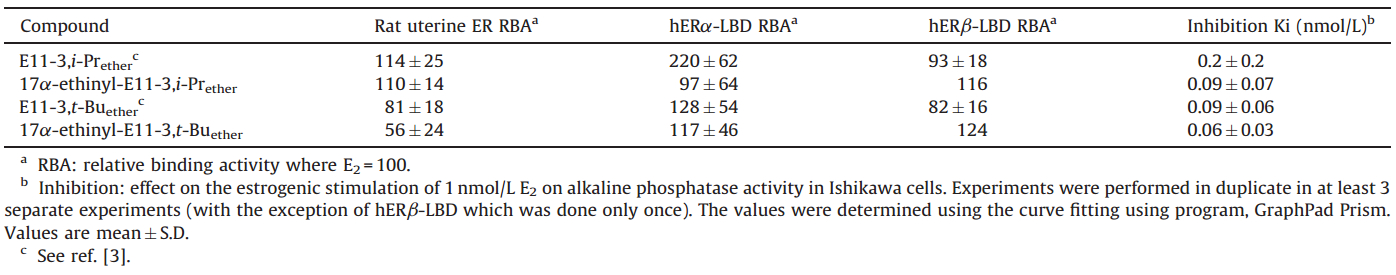
In recent years estrogen receptor antagonists have become very important therapeutic agents for the treatment of estrogen sensitive cancers,such as breast cancer [1]. Many anti-estrogenic compounds are typified by analogs of estradiol,substituted at C-7a (ICI 164,384) and C-11b(RU39411) or aromatic compounds,such as Tamoxifen. They share some similar structural features,mainly polar long chains containing tertiary amines or carboxylic groups (Fig. 1).

|
Download:
|
| Fig. 1.The structures of the classical antiestrogens, raloxifene, tamoxifen, RU 39411; the estrogen E11-2,1 and the antiestrogens E11-2,2 and E11-2,2ether. | |
We have identified a series of unusual estradiol analogs with short and non-polar substituent groups at the 11bposition [2, 3]. One of the most remarkable examples is the simple and short chain ester,E11-2,2 (Fig. 1). The highly unusual aspect of these results is that a complete reversal of function occurs with a single methylene group,lengthening the side-chain from 4 atoms of a methyl ester in E11-2,1 to 5 atoms of an ethyl ester in E11-2,2 (Fig. 1). E11-2,1 is an estrogen receptor agonist,while E11-2,2 an antagonist. This same principle was found to apply to other substitutions at the 11b position,which include ketones,ethers (Fig. 1,E11-2,2ether) and thiono esters [3].
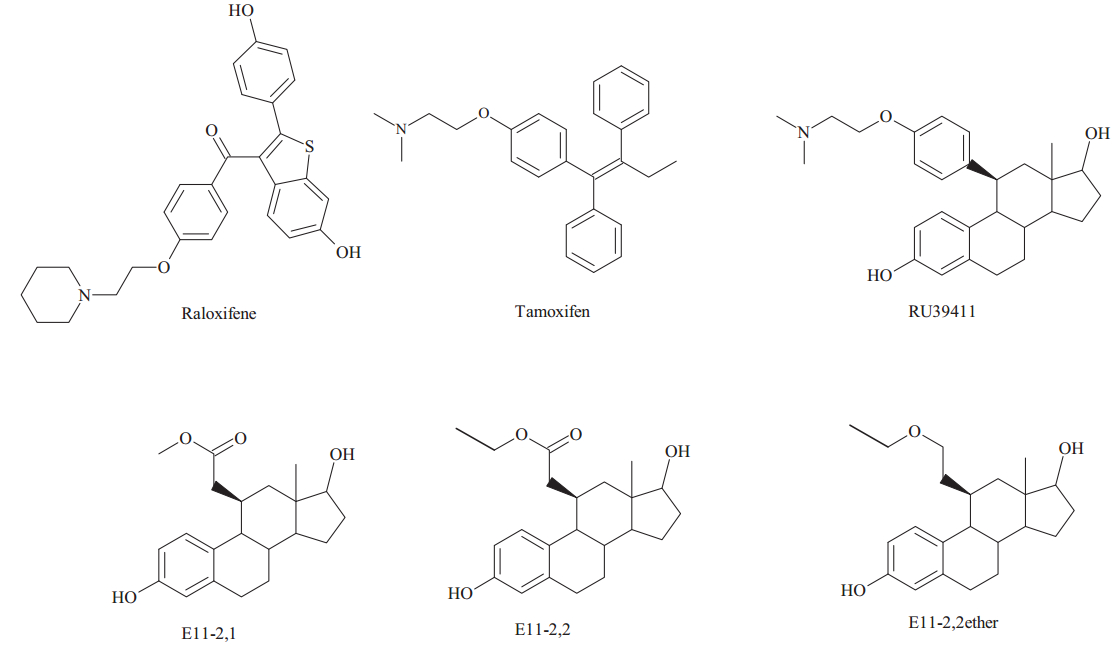
The ethers were especially interesting as they were highly active anti-estrogens and were more stable than other substituents [3]. Among these ethers,two of them,11b-(3-isopropoxypropyl)-estra-1,3,5(10)-trien-3,17β-diol and 11b-(3-t-butoxypropyl)estra-1,3,5(10)-trien-3,17β-diol (E11-3,i-Pretherand E11-3,t-Buether) were especially potent anti-estrogens that were totally without agonist activity. Consequently,in order to produce an ER antagonist that would be longer lived and could be administered orally,we introduced an ethinyl group at 17αposition of estradiol to render the D-ring of the steroid resistant to oxidation [4]. These compounds are analogous to moxestrol,11b-methoxy-17α-ethinyl-estradiol,an extremely potent estrogen that is highly resistant to metabolism [5]. The synthesis and biological activity of these two compounds 11b-(3-isopropoxypropyl)estra-17α-ethinyl-1,3,5(10)-trien-3,17β-diol and 11b-(3-t-butoxypropyl)estra-17α-ethinyl-1,3,5(10)-trien-3,17β-diol (17α-ethinylE11-3,i-Pretherand 17α-ethinyl-E11-3,t-Buether,Fig. 2) are reported here.

|
Download:
|
| Fig. 2. The structures of 17α-ethinyl-E11-3,i-Prether and 17α-ethinyl-E11-3,t-Buether. | |
General:1H NMR spectra were recorded with a Bruker Avance 400 spectrometer,and chemical shifts are reported relative to residual CHCl3(7.27 ppm). Purification by flash chromatography was performed according to the procedure of Still [6],using 230-400 mesh silica gel (EM Science,Darmstadt Germany). Unless otherwise noted,solvents (analytical or HPLC) and reagents were used as supplied,and all reactions were carried out under nitrogen. Thin-layer chromatography (TLC) was performed using Merck silica gel plates (F254) (EM Science) and visualized using phosphomolybdic acid or UV illumination. TL system: T-1,hexanes/EtOAc (2:1); T-2,hexanes/EtOAc (1:1). Analytical high performance liquid chromatography (HPLC) was performed on a Beckman System Gold HPLC system (Beckman Coulter,Inc.,Fullerton,CA) consisting of a model 126 solvent module and a model 168 diode array detector at 280 nm using the following columns and systems: with an RP-18 column (LiChrosorb RP-18,5mm,4.6 mm X 25 cm,EM Science),with H-1, CH3CN/H2O (50/50) at 3 mL/min,280 nm; with a Diol column (LiChrosphor Diol,5mm,4.6 mm X 25 cm,EM Science) with,H-2, CH2Cl2 at 1 mL/min,280 nm.
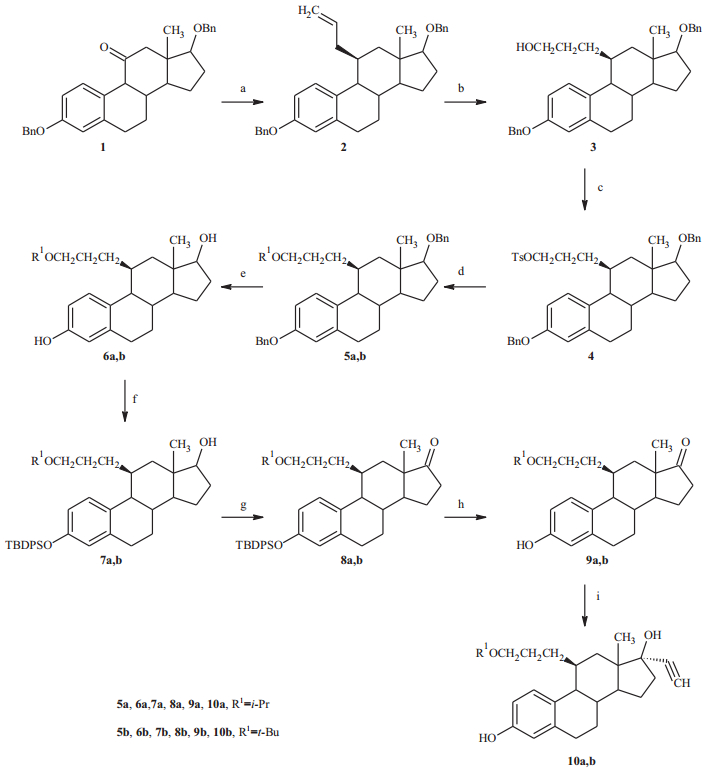
Synthesis of 3-hydroxy,17β-hydroxy,17α-ethinyl,11β-substituted estradiols is shown in Scheme 1.

|
Download:
|
| Scheme 1.Reagents and conditions: (a): (i) allylmagnesium bromide, THF, (ii) HSiEt3,BF3.Et2O, 0℃(1–2). (b): (i) catecholborane, LiBH4, THF, (ii) NaOH, H2O2(2–3). (c) TsCl,Pyr, 0℃(3–4). (d): (i) KH, 18-crown-6, ROH, toluene, r.t., (ii)4,80℃(4–5a); (iii)t-BuOK (4–5b). (e) 5% Pd-C, H2, EtOAc-EtOH, r.t. (5–6). (f)t-BuDPSO, Et3N, 4-Me2N-pyridine,CH2Cl2, r.t. (6–7). (g) pyridinium chlorochromate (PCC), NaOAc, CH2Cl2, r.t. (7–8). (h) tetrabutylammonium fluroride, THF, r.t. (8–9). (I) (i) 18% sodium acetylide, xylene, r.t., (ii)NH4Cl (9–10). | |
The synthesis of 3,17β-dibenzyloxyestra-1,3,5(10)-trien-11-one (1) was prepared as previously described in the literature [7]. Compound1was first converted to 11a-allyl-3,17β-dibenzyloxyestra-1,3,5(10)-triene-11b-ol by addition of allylmagnesium bromide. Ally group was introduced from the less sterically hindered alpha side. The hydroxyl group was then reduced by triethylsilane and BF3·Et2O at 0℃ to yc,17β-dibenzyloxy estra-1,3,5(10)-triene (2),in which the configuration of 11-ally was inversed from 11a to 11b. Hydroboration of the terminal olefin with LiBH4and catecholborane followed by oxidation with H2O2 resulted in propanol compound 3[8]. Tosylation of the hydroxyl group by p-toluenesulfonyl chloride in pyridine gave compound4,which was converted to the desired ethers in the next step. To generate the 3,17-bis-protected E11-3,i-Prether (5a),isopropanol was first activated by reacting with a suspension of 35% dispersion of KH in the presence of 18-crown-6. Reacting with this anion of isopropanol in toluene at 80℃ converted tosylate (4)to the protected ether (5a),which was then deprotected by hydrogenolysis of the benzyl groups using 5% palladium on carbon under an atmosphere of H2to give 6a.
The 3,17-bis-protected E11-3,t-Buether(5b) was prepared from the protected tosylate (4) using solid potassium t-butoxide instead of KH and isopropanol. Deprotection and purification as above gave 6b.
To prepare 17α-ethinyl E11-3,i-Prether,another strategy of protection/deprotection was employed. The 3-hydroxy group was selectively protected byt-butyldiphenylsilyl chloride in CH2Cl2 in the presence of 4-(dimethylamino)pyridine and triethylamine to give the 3-protected ether (7a). The 17-hydroxy group was then oxidized by pyridinium chlorochromate and sodium acetate in CH2Cl2 to give the 3-protected-17-keton (8a). Deprotection of 3-hyroxygroup by tetrabutylammonium fluroride in THF gave the 3-hydroxy-17-keton (9a).
Finally,to obtain 17α-ethinyl E11-3,i-Prether(10a),a solution of 18% sodium acetylide in xylenes was first added to the above deprotected ketone (9a) in DMSO. After reacting at r.t. for 3.5 h,the reaction was poured into saturated aqueous NH4Cl and extracted with EtOAc. Flash chromatography gave the 17α-ethinyl E11-3,i-Prether (10a). 17α-ethinyl E11-3,t-Buether (10b) was similarly prepared according to the above procedures.
Detailed experimental procedures and data for compounds7a, 8a,9a,10aand10bare listed in ref. [12]. 2.2. Biological studies
The binding of the two 11β-substituted ethers to rat uterine cytosol ER,human Estrogen Receptoraligand Binding Domain (hERa-LBD) and human Estrogen Receptor β ligand Binding Domain (hERβ-LBD) [9] were determined by competition for the binding of 1 nmol/L [3H] E2as previously described [3]. Relative binding affinity (RBA) was determined by analysis of the displacement curves by the curve-fitting program Prism. The results as RBAs compared to E2represent the ratio of the EC50of E2 to that of the steroid analogs X 100.
The anti-estrogenic potency of the two 11 β-substituted ethers were determined by the inhibition of the effect of 1 nmol/L E2on the estrogen sensitive marker,alkaline Phosphatase,in Ishikawa cells [10] using the procedure previously described [3]. For antagonists,the effect (Ki) of each compound tested at a range from 10-6 to 10-12 mol/L was measured for the inhibition of the action of 10-9 mol/L E2(EC50= 0.2 nmol/L). Each compound was analyzed in at least three separate experiments performed in duplicate. The Ki were determined using the curve fitting program Prism. 3. Results and discussion
The biological properties of the two 11 β-ethers substituted with a 17α-ethinyl group were analyzed in several different assays: The affinity of the compounds for the estrogen receptor (ER) was determined by their competition for the binding of [3H]E2: rat uterine cytosol (native ER,predominantly ERa[11]); ligand binding domain (LBD) of human ERaand the LBD of human ERβ. Anti-estrogenic potency was measured by inhibition of the stimulation of alkaline phosphatase in Ishikawa cells by 1 nmol/L E2. The results summarized in Table 1 are compared to the previously reported results [3] of the parent compounds (unsubstituted at 17α). As shown in Table 1,these two compounds,17αethinyl-E11-3,t-Buether and 17α-ethinyl-E11-3,i-Prether,bindvery strongly to both ERaand ERβ,with relative binding affinities approximately equal to that of E2. In each case,the 17α-ethinyl substituted compounds bound equally,or slightly weaker,than the parents. More importantly,both compounds are extremely potent anti-estrogens with potencies equal to,or greater than,that of the unsubstituted parent compounds; most likely reflecting their resistance to metabolism in the cell. Thus,it is highly likely that both compounds are promising for antiestrogen drug development.
|
|
{kind=link}
{kind=link}
{kind=link}
We have generated two estradiols with 11b-i-PrO-propyl and 11b-t-BuO-propyl ethers substitutions and demonstrated their extremely strong antagonist activities.
| [1] | S.X. Lin, J. Chen, M. Mazumdar, et al., Molecular therapy of breast cancer: progress and future directions, Nat. Rev. Endocrinol. 6 (2010) 485-493. |
| [2] | J.X. Zhang, D.C. Labaree, G. Mor, et al., Estrogen to antiestrogen with a single methylene group resulting in an unusual steroidal selective estrogen receptor modulator, J. Clin. Endocrinol. Metab. 89 (2004) 3527-3535. |
| [3] | J.X. Zhang, D.C. Labaree, R.B. Hochberg, Nonpolar and short side chain groups at C-11b of estradiol result in antiestrogens, J. Med. Chem. 48 (2005) 1428-1447. |
| [4] | S.M. Hyder, C. Chiappetta, G.M. Stancel, Synthetic estrogen 17α-ethinyl estradiol induces pattern of uterine gene expression similar to endogenous estrogen, J. Pharmacol. Exp. Ther. 290 (1990) 740-747. |
| [5] | J. Salmon, D. Coussediere, C. Cousty, J.P. Raynaud, Pharmaco kinetics and metabolism of moxestrol in animals (rat, dog, monkey), J. Ster. Biochem. 19 (1983) 1223-1234. |
| [6] | W.C. Still, M. Kahn, A. Mitra, Rapid chromatographic technique for preparative separations with moderate resolution, J. Org. Chem. 43 (1978) 2923-2925. |
| [7] | D.C. Labaree, J.X. Zhang, H.A. Harris, et al., Synthesis and evaluation of B-, C-, and D-ring-substituted estradiol carboxylic acid esters as locally active estrogens, J. Med. Chem. 46 (2003) 1886-1904. |
| [8] | R. Tedesco, R. Fiaschi, E. Napolitano, Novel stereoselective synthesis of 11 b-carbon-substituted estradiol derivatives, J. Org. Chem. 60 (1995) 5316-5318. |
| [9] | H.A. Harris, A.R. Bapat, D.S. Gonder, D.E. Frail, The ligand binding profiles of estrogen receptors alpha and beta are species dependent, Steroids 67 (2002) 379-384. |
| [10] | B.A. Littlefield,E.Gurpide,L.Markiewicz, et al.,Asimpleandsensitivemicrotiterplate estrogen bioassay based on stimulation of alkaline phosphatase in Ishikawa cells: estrogenic action of delta 5 adrenal steroids, Endocrinology 127 (1990) 2757-2762. |
| [11] | G.G. Kuiper, B. Carlsson, K. Grandien, et al., Comparison of the ligand binding specificity and transcript tissue distribution of estrogen receptors alpha and beta, Endocrinology 138 (1997) 863-870. |
| [12] | Detailed experimental procedures and data for compounds 7a, 8a, 9a, 10a and 10b:3-t-Butyldiphenylsiloxy-11β-(3-isopropoxypropyl)estra-1,3,5(10)-trien- 17β-ol (7a). A solution of 16 mg (0.0432 mmol) of E11-3,i-Prether (6a), t-butyldiphenylsilyl chloride (124 mL, 0.475 mmol), dimethylaminopyridine (10 mg, 0.08 mmol) and triethylamine (200 mL, 1.434 mmol) in CH2Cl2 (1 mL) was allowed to stir at r.t. overnight. The reaction was poured into H2O (50 mL) and extracted with EtOAc (3×50 mL). Combined organic extracts were dried over Na2SO4 and concentrated in vacuo giving a clear colorless oil. Purification by flash chromatography on a 2 cm×17 cm column of silica gel using 2:1 hexanes/EtOAc as eluent gave 14.4 mg (54%) of 7a. Data for 7a: TLC, T-1, Rf 0.35.3-t-Butyldiphenylsiloxy- 11β-(3-isopropoxypropyl)estra-1,3,5(10)-trien-17-one (8a). A solution of 7a, NaOAc (1 mg) in CH2Cl2 (2 mL) was stirred at r.t. as PCC (8 mg, 0.035 mmol) was added. The reaction was stirred at r.t. for 2 h, poured into H2O (30 mL) and extracted with EtOAc (3×20 mL). Combined organic extracts were dried over Na2SO4 and concentrated in vacuo giving a brown film. Purification by flash chromatography on a 2 17 cm column of silica gel using 4:1 hexanes/EtOAc as eluent gave 10.3 mg (71%) of 8a. Data for 8a: TLC, T-1, Rf 0.55.3-Hydroxy-11β- (3-isopropoxypropyl)estra-1,3,5(10)-trien-17-one (9a). A solution of 1 mol/L tetra-n-butylammonium fluoride in THF (1 mL) was added to 8a (10.3 mg, 0.0169 mmol) and stirred at r.t. for 1.5 h. The reaction was poured into H2O (70 mL) and extracted with EtOAc (3×20 mL). Combined organic extracts were dried over Na2SO4 and concentrated in vacuo giving a clear colorless oil. Purification by flash chromatography on a 2 cm×17 cm column of silica gel using 2:1 hexanes/EtOAc as eluent gave 5 mg (79%) of 9a. Data for 9a: TLC, T-1, Rf 3.75; 1H NMR (400 MHz, CDCl3): d 1.05 (s, 3H, H-18), 1.11 (d, 3H, J = 6.1 Hz, -CH3), 1.12 (d, 3H, J = 6.1 Hz, -CH3), 2.18 (dd, 1H, J = 13.9, 1.6 Hz, H-12b), 2.50-2.55 (m, 1H, H- 11), 2.54 (dd, 1H, J = 18.9, 8.6 Hz, H-16b), 2.60 (dd, 1H, J = 10.9, 4.6 Hz, H-9), 2.72- 2.87 (m, 2H, H-6), 3.28-3.37 (m, 2H, CH2O), 3.51 (septet, 1H, J = 6.1 Hz, CH(CH3)2), 4.70 (br s, 1H, OH), 6.56 (d, 1H, J = 2.7 Hz, H-4), 6.65 (dd, 1H, J = 8.5, 2.7 Hz, H-2), 7.04 (d, 1H, J = 8.5 Hz, H-1).11β-(3-Isopropoxypropyl)estra-17α-ethinyl- 1,3,5(10)-trien-3,17β-diol (10a). A solution of 9a (5 mg) in DMSO (2 mL) was stirred at r.t. as an 18% solution of sodium acetylide in xylene/mineral oil (1 mL) was added over 5 min. The reaction was stirred at r.t. for 3.5 h, poured into saturated aqueous NH4Cl (30 mL) and extracted with EtOAc (3×30 mL). Combined organic extracts were dried over Na2SO4 and concentrated in vacuo giving a yellow oil (DMSO was azeotroped off with toluene). Purification by flash chromatography on a 1 cm×17 cm column of silica gel using 1:1 hexanes/EtOAc as eluent gave product contaminated with a nonpolar impurity. Further purification in 6 portions by semiprep HPLC (RP-18) eluting at 3 mL/min with 50/50 CH3CN/ H2O (tR, 15 min) followed by crystallization from acetone-petroleum ether gave 3.4 mg (63%) of 10a. Data for 10a: TLC, T-1, 0.35; 1HNMR(400 MHz, CDCl3): d 1.04 (s, 3H, H-18), 1.12 (d, 3H, J = 6.1 Hz, -CH3), 1.13 (d, 3H, J = 6.1 Hz, -CH3), 2.51-2.56 (m, 1H, H-11), 2.60 (dd, 1H, J = 10.5, 4.3 Hz, H-9), 2.64 (s, 1H, ethinyl-H), 2.67-2.83 (m, 2H, H-6), 3.32 (t, 2H, J = 6.9 Hz, CH2O), 3.52 (septet, 1H, J = 6.1 Hz, -CH(CH3)2), 6.54 (d, 1H, J = 2.7 Hz, H-4), 6.64 (dd, 1H, J = 8.5, 2.7 Hz, H-2), 7.05 (d, J = 8.5 Hz, H- 1); HPLC system: H-2, tR = 11.16 min; system H-1, tR 15 min, >99% pure.11β-(3- t-Butoxypropyl)estra-17α-ethinyl-1,3,5(10)-trien-3,17β-diol (10b). A solution of 9b (5.3 mg, 0.01378 mmol) inDMSO (2 mL) was stirred at r.t. as an 18% solution of sodium acetylide in xylene/mineral oil (2 mL) was added dropwise over 5 min. The reaction was stirred at r.t. for 3.5 h, poured into saturated aqueous NH4Cl (60 mL) and extracted with EtOAc (2×50 mL). Combined organic extracts were dried over Na2SO4 and concentrated in |