




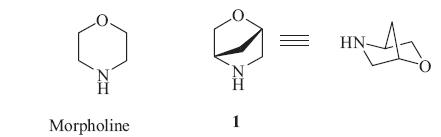
Bridged bicyclic morpholines have been developed as morpholine isosteres due to their similarity in structure and properties such as lipophilicity [1]. The bicyclic morpholine (1S,4S)-2-oxa-5- azabicyclo[2.2.1]heptane (1) has been utilized successfully as a morpholine surrogate in drug design [2]. Given the structural similarity between 1 and morpholine (Fig. 1) and potential similarities in physicochemical and pharmacokinetic property of compounds containing 1 moiety compared to those of morpholine derivatives,it is anticipated that this bicyclic morpholine 1 will become an important building block in medicinal chemistry research.

|
Download:
|
| Fig. 1.Morpholine and bridged bicyclic morpholine: (1S,4S)-2-oxa-5- azabicyclo[2.2.1]heptane (1). | |
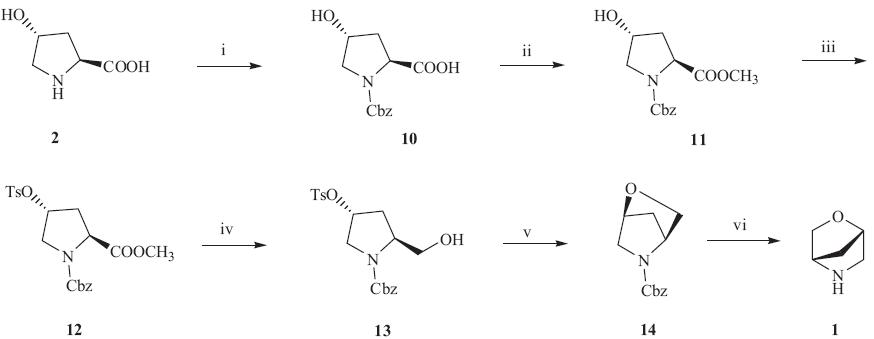
Despite the potential of bridged bicyclic morpholine 1 to become a useful morpholine isostere,to the best of our knowledge, only two synthetic routes with the same strategy have been reported on the synthesis of 1,using trans-4-hydroxy-L-proline (2) as the starting material with the main difference on the Nprotection group [3, 4, 5]. The first synthesis was reported by Portoghese in 1971 using N-benzoyl protected group,with a total yield of 59% in a seven-step procedure [3, 4]. After that,in Sun’s method in 2006,Boc group was utilized in a six-step procedure but without yields reported [5]. Although Portoghese’s protocol (Scheme 1) was in good total yield,the deprotection of benzoyl group seemed to be tedious,and using diazomethane to generate methyl ester 4 is toxic and dangerous. Keeping these aspects in mind,efforts were focused on developing a facile and green synthetic approach of this bridged morpholine from the cheap, commercially available trans-4-hydroxy-L-proline (2) as the starting material.

|
Download:
|
| Scheme 1.Portoghese’s synthesis of (1S,4S)-2-oxa-5-azabicyclo[2.2.1]heptane hydrochloride (9). Reagents and conditions: (a) NaOH,benzoyl chloride,Et2O,r.t.,24 h,90%; (b) CH2N2,MeOH,r.t.,98%; (c) TsCl,Py,5 °C,60 h,94%; (d) LiBH4,1,2-dimethoxyethane,0 °C to r.t.,4 h,93%; (e) NaOMe,MeOH,EtOH,reflux,2 h,90%; (f) BH3/THF,0 °C to reflux,1 h,100%; (g) 10% Pd/C,H2,2 mol/L HCl,85%. | |
Encouraged by the promising result of almost quantitative yield from trans-4-hydroxy-L-proline (2) to N-benzyloxycarbonyl-trans- 4-hydroxy-L-proline and benzyloxycarbonyl (Cbz) protecting group can be removed smoothly by catalytic hydrogenation [6], herein,we report a facile six-step synthesis of (1S,4S)-2-oxa-5- azabicyclo[2.2.1]heptane (1) in excellent total yield of 70% using NCbz protected protocol (Scheme 2).

|
Download:
|
| Scheme 2.Synthesis of (lS,4S)-2-oxa-5-azabicyclo[2.2.1]heptane (1). Reagents and conditions: (i) NaOH,CbzCl,H2O,0 °C to r.t.,5 h,91%; (ii) SOCl2,MeOH,0 °C to r.t.,2.5 h, 96%; (iii) TsCl,DMAP,Et3N,CH2Cl2,0 °C to r.t.,15 h,93%; (iv) NaBH4,EtOH/THF,0 °C to r.t.,16 h,100%; (v) NaOMe,MeOH,reflux,4 h,86%; (vi) 10% Pd/C,H2,MeOH,4 h,100%. | |
The 1H NMR and 13C NMR spectra were recorded in CDCl3 solution on a Varian mercury-400 at 400 and 100 MHz respectively. 1H NMR and 13C NMR spectra were reported in parts per million using TMS (δ 0.00) as an internal standard. High-resolution mass spectra were measured on a Theromo Extractive Orbitrap plus mass spectrometer (ESI). The optical rotation was measured on a PerkinElmer 241MC polarimeter. All of the reagents and solvents were purchased from commercial sources.
Typical procedure for the preparation of (2S,4R)-1-benzyloxycarbonyl- 4-hydroxypyrrolidine-2-carboxylic acid (10): A solution of 2 (6.56 g,0.05 mol) in H2O (50 mL) was added NaOH (4 g, 0.1 mol) at 0 °C. Benzyl chloroformate (8.5 g,0.05 mol) was then added dropwise at 0 °C and stirred for 5 h at room temperature. The reaction mixture was extracted with ether (20 mL),and the aqueous layer was neutralized with 2 mol/L HCl to pH 2,and extracted with ethyl acetate (30 mL × 3). The organic layer was combined,washed with brine,dried over Na2SO4,and concentrated under vacuum to give the target compound (12.1 g,yield: 91%).The 1H NMR spectra were in accordance with the literature data [7]. 1H NMR (400 MHz,CDCl3): δ 7.27–7.32 (m,5 H),6.17 (brs,1 H), 5.09–5.12 (m,2 H),4.45–4.51 (m,2 H),3.60 (brs,2 H),2.14–2.30 (m,2 H).
Typical procedure for the preparation of methyl (2S,4R)-1- benzyloxycarbonyl-4-hydroxypyrrolidine-2-carboxylate (11): SOCl2 (3.83 g,32.2 mmol) was added to a solution of 10 (7.78 g, 29.3 mmol) in CH3OH (25 mL) dropwise at 0 °C,and the mixture was stirred for 2.5 h at room temperature. The solvent was concentrated under vacuum to afford a viscous oil (7.86 g,yield: 96%). The 1H NMR spectra were in accordance with the literature data [8]. 1HNMR(400 MHz,CDCl3): δ 7.30–7.35 (m,5 H),5.09–5.21 (m,2 H),4.50–4.54 (m,2 H),3.75 and 3.54 (each s,total 3 H),3.65– 3.70 (m,2 H),2.25–2.35 (m,1 H),2.05–2.18 (m,2 H).
Typical procedure for the preparation of methyl (2S,4R)-1- benzyloxycarbonyl-4-(tosyloxy)pyrrolidine-2-carboxylate (12): A solution of compound 11 (2.22 g,7.95 mmol),Et3N (0.88 g, 8.7 mmol),and DMAP (0.97 g,7.95 mmol) in CH2Cl2 (30 mL) was cooled to 0 °C. Then,p-toluenesulfonyl chloride (1.67 g, 8.7 mmol) was added in one portion and stirred for 15 h at room temperature. The mixture was washed with water and brine,dried over Na2SO4 and concentrated under vacuum. The residue was purified by flash column chromatography (EA in PE: 30–40%) to give a colorless oil (3.2 g,yield: 93%). The 1H NMR spectra were in accordance with the literature data [9]. 1H NMR (400 MHz,CDCl3): δ 7.76 (d,2 H,J = 8.0 Hz),7.29–7.37 (m,7 H),4.99–5.20 (m,3 H), 4.45–4.48 (m,1 H),3.53 and 3.75 (each s,total 3 H),3.63–3.72 (m,2 H),2.55–2.60 (m,1 H),2.43 and 2.46 (each s,total 3 H),2.16–2.21 (m,1 H).
Typical procedure for the preparation of (2S,4R)-1-benzyloxycarbonyl- 2-hydroxymethyl-4-(tosyloxy)pyrrolidine (13): NaBH4 (2.27 g,60 mmol) was added to a solution of 12 (4.33 g,10 mmol) in ethanol (50 mL) and THF (30 mL) in one portion at 0 °C. The mixture was stirred for 16 h at room temperature. Glacial acetic acid (4.25 mL) was added carefully at 0 °C,and then the mixture was evaporated to remove the solvent. Water and ethyl acetate were added to the residue. The aqueous layer was extracted with ethyl acetate (30 mL × 3),while the organic layer was combined, washed with brine,dried over Na2SO4,and concentrated under vacuum to give the target compound (4.05 g,yield: 100%). The 1H NMR spectra were in accordance with the literature data [10]. 1H NMR (400 MHz,CDCl3):δ 7.75–7.77 (m,2 H),7.32–7.39 (m,7 H),5.08–5.15 (m,2 H),5.02 (brs,1 H),4.09–4.13 (m,1 H), 3.77–3.83 (m,2 H),3.50–3.57 (m,2 H),2.44 (s,3 H),2.19–2.24 (m,1 H),1.78–1.84 (m,1 H).
Typical procedure for the preparation of (1S,4S)-5-benzyloxycarbonyl- 2-oxa-5-azabicyclo[2.2.1]heptane (14): A mixture of 13 (4.05 g,10 mmol) and NaOMe (0.81 g,15 mmol) in dry CH3OH (20 mL) was refluxed for 4 h,then cooled to room temperature. The solvent was removed under vacuum,followed by addition of sat. Na2CO3 (100 mL). The mixture was extracted with ethyl acetate (30 mL × 3),and the organic layer was combined,washed with brine,dried over Na2SO4,and purified by flash column chromatography (EA in PE: 30–50%) to give a colorless oil (2.01 g,yield: 86%). 1H NMR (400 MHz,CDCl3): δ 7.33–7.35 (m,5 H),5.13–5.15 (m,2 H),4.54–4.61 (m,2 H),3.70–3.92 (m,2 H),3.43–3.47 (m,1 H), 3.34–3.39 (m,1 H),1.81–1.88 (m,2 H). 13C NMR (100 MHz,CDCl3): (rotamers) δ 154.5,136.6,128.4,128.0,127.9,127.7,76.3/76.0, 74.2,66.9/66.7,57.2/56.9,54.7/54.5,36.6/36.1. HRMS (ESI-TOF+) Calcd. for C13H16NO3 [M+H]+: m/z 234.1125; found,234.1125. [α]20D =-21:3(c1:6; CHCl3).
Typical procedure for the preparation of (1S,4S)-2-oxa-5- azabicyclo[2.2.1]heptane (1): To a solution of 14 (1.16 g,5 mmol) in CH3OH (20 mL) was added 10% Pd/C (0.12 g). The mixture was stirred under H2 at atmospheric pressure for 4 h at room temperature. After filtration,the filtrate was evaporated in vacuo at 35 °C to give the title compound (0.5 g,yield: 100%). 1H NMR (400 MHz,CDCl3): δ 4.50 (s,1 H),3.81 (d,1 H,J = 7.2 Hz),3.73–3.75 (m,2 H),3.04–3.07 (m,2 H),2.96 (d,1 H,J = 10.4 Hz),1.80 (d,1 H, J = 9.6 Hz),1.73 (d,1 H,J = 9.6 Hz). 13C NMR (100 MHz,CDCl3): δ 76.2,75.9,56.3,52.8,36.3. HRMS (ESI-TOF+) Calcd. for C5H10NO: m/z 100.0757; found: 100.0760. [α]20 D +=104.2(c1:25; CHCl3). 3. Results and discussion
The title compound was synthesized by a convenient six-step procedure as outlined in Scheme 2. In our strategy,the critical step is to choose benzyloxycarbonyl (Cbz) as the protection group of amino in trans-4-hydroxy-L-proline (2). Treatment of 2 with benzylchloroformate (Cbz-Cl) in the presence of NaOH afforded carbamate 10 conveniently in 91% yield [6]. Moreover,instead of using toxic and dangerous diazomethane,MeOH/SOCl2 was employed to make the methyl ester 11 mildly [8]. Several conditions were also screened for the synthesis of tosylate 12. The results were summarized in Table 1. It was found that using triethylamine as the base and one equivalent of DMAP as a catalyst was beneficial to promote the transformation at room temperature in terms of reaction time and yield. Reduction of methyl ester to alcohol proceeded smoothly by treatment with NaBH4 in EtOH/ THF,giving alcohol 13 quantitatively. The use of NaBH4 instead of LiBH4 lowered the cost,which was another advantage compared to Portoghese’s protocol [3]. The formation of the desired bridged compound 14 was achieved by treatment of the alcohol 13 in good yield in a solution of NaOMe in MeOH. Finally,the Cbz protecting group in 14 was removed smoothly by catalytic hydrogenation in the presence of catalytic amount of Pd/C to afford the bridged morpholine 1 in quantitative yield. The total yield was 70%. Overall, this synthetic route was efficient and easy to scale-up.
| Table 1 The exploration of reaction conditions for the synthesis of tosylate 12. |

{kind=link}
{kind=link}
{kind=link}
In summary,a short,efficient,and facile synthesis of (1S,4S)-2- oxa-5-azabicyclo[2.2.1]heptane (1) was accomplished in six steps in an excellent yield of 70% starting from commercially available and inexpensive trans-4-hydroxy-L-proline (2). This improved synthetic method of bridged morpholine 1 will make morpholine isostere more attractive in medicinal chemistry research.
| [1] | D.G. Wishka, P. Beagley, J. Lyon, et al., A concise synthesis of 6-oxa-3-azabicyclo[3.1.1]heptane hydrotosylate, Synthesis 16 (2011) 2619-2624. |
| [2] | A.J. Roecker, P.J. Coleman, S.P. Mercer, et al., Kinesin spindle protein (KSP) inhibitors. Part 8: Design and synthesis of 1,4-diaryl-4,5-dihydropyrazoles as potent inhibitors of the mitotic kinesin KSP, Bioorg. Med. Chem. Lett. 17 (2007) 5677-5682. |
| [3] | P.S. Portoghese, J.G. Turcotte, Synthesis and nuclear magnetic resonance investigation of some chiral 2-oxa-5-azabicyclo[2.2.1]heptane derivatives, Tetrahedron 27 (1971) 961-967. |
| [4] | P.S. Portoghese, J.G. Turcotte, Stereochemical studies on medicinal agents. 9. Bicyclic bases. Synthesis and biological activities of epimeric quaternary derivatives of 2-oxa-5-azabicyclo[2.2.1]heptane, J. Med. Chem. 14 (1971) 288-291. |
| [5] | P.Y. Sun, Quinazoline compounds or their medical salts and preparation and medical usage thereof, WO2006081741. |
| [6] | R. Ganorkar, A. Natarajan, A. Mamai, J.S. Madalengoitia, Synthesis of conformationally constrained lysine analogues, J. Org. Chem. 71 (2006) 5004-5010. |
| [7] | F. Fache, O. Piva, Synthesis and applications of the first polyfluorous proline derivative, Tetrahedron: Asymmetry 14 (2003) 139-143. |
| [8] | S.C. Mayer, J. Ramanjulu, M.D. Vera, A.J. Pfizenmayer, M.M. Joullié, Synthesis of new didemnin B analogs for investigations of structure/biological activity relationships, J. Org. Chem. 59 (1994) 5192-5205. |
| [9] | J. Chiba, G. Takayama, T. Takashi, et al., Synthesis, biological evaluation, and pharmacokinetic study of prolyl-1-piperazinylacetic acid and prolyl-4-piperidinylacetic acid derivatives as VLA-4 antagonists, Bioorg. Med. Chem. 14 (2006) 2725-2746. |
| [10] | F.M. Kaspersen, U.K. Pandit, Unconventional nucleotide analogues. Part XⅢ. (2S,4S)-2-hydroxymethyl-and 2-carboxy-4-(purin-9-yl) pyrrolidines, J. Chem. Soc. Perkin Trans. 1 (1975) 1617-1622. |