




b School of Clinical Medicine, Wenzhou Medical University, Wenzhou 325035, China
Pharmacological targeting of proteins that regulate epigenetics has emerged as a promising therapeutic area of study [1, 2]. Histone acetylation and deacetylation play a crucial role for regulating protein function of eukaryotic cells,which is correlated with two classes of enzymes: histone deacetylases (HDACs) and histone acetyltransferases (HATs) [3]. The maintenance of equilibrium between acetylation and deacetylation of histones and nonhistone substrates is essential for normal cell growth. However, HDAC overexpression has been found in a variety of human cancers,including myeloid neoplasia and solid tumors [4]. Therefore,HDACs represent a rational target for intervention of a number of cancers,and the development of histone deacetylase inhibitors (HDACi) has become an efficacious therapeutic strategy in cancer treatment [5, 6, 7].
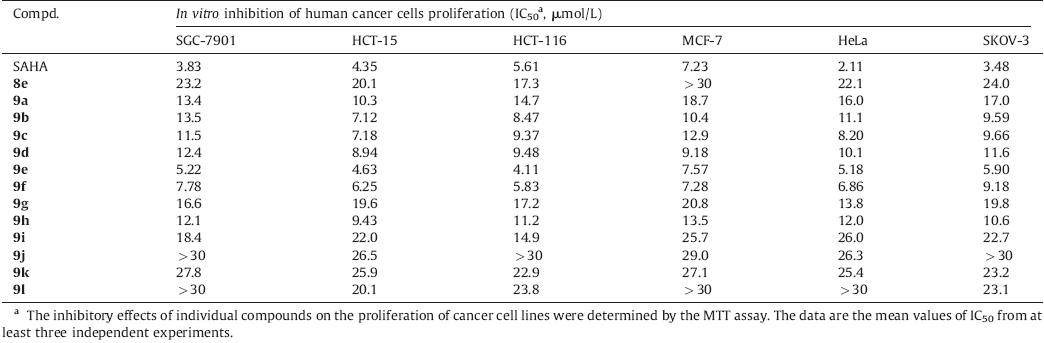
There are a number of HDACi which are currently undergoing clinical trials,either mono-therapy or combination therapy,for cancer treatment (Fig. 1). Most of HDACi are pan-inhibitors,but there are also isoform or class selective HDACi in the clinical development stage [8, 9]. More importantly,vorinostat (SAHA) and romidepsin (FK228),as the verified HDACi,have been approved by the FDA for the treatment of cutaneous T-cell lymphoma (CTCL) [10, 11]. These two approved drugs have validated the therapeutic use of HDACi in cancer therapy. Other compounds in clinical trials [12, 13, 14, 15, 16, 17] for the treatment of hematological and solid tumors include LBH589 (panobinostat,Novartis,phase III),PXD101 (belinostat,Topotarget,phase III),SB939 (pracinostat,S*BIO,phase II),TSA (trichostatin A),MS-275 (entinostat,Syndax,phase II),and MGCD-0103 (mocetinostat,Methylgene,phase II) (Fig. 1). Among the approved HDACi or in clinical trials,LBH589,PXD101,and SB- 939 share a common active fragment,N-hydroxycinnamamide. This fragment could not only undergo bichelation with the active site Zn2 + by its hydroxamic acid group,but also form a sandwichlike π-π interaction by inserting its vinyl benzene group into two parallel phenylalanine residues of HDAC [9, 18].

|
Download:
|
| Fig. 1.Approved and clinical trial HDAC inhibitors. | |
Melting points were determined on a RDCSY-I capillary apparatus and were uncorrected. The compounds synthesized were purified by column chromatography using silica gel (200– 300 mesh) except for recrystallization and thin-layer chromatography (TLC) using silica gel 60 F254 plates (250 mm; Qingdao Ocean Chemical Company,China). The 1H NMR spectra were recorded with a Bruker Avance 300 MHz spectrometer at 300 K,using TMS as an internal standard. MS spectra were recorded on a Mariner Mass Spectrum (ESI). Element analyses were performed on an Eager 300 instrument.
The synthetic route of these target compounds is outlined in Scheme 1. The carboxyl group of FA 1 was protected to form methyl ferulicate 2 using SOCl2 in MeOH solution. Then,compound 2 was reacted with various dibromoalkanes to obtain compounds 3a–c>. Subsequently,3a–c were ammoniated with ammonium hydroxide or alkylamine to give key intermediates 4a–l. In addition, acetylsalicylic acid 5 was treated with oxalylchloride to produce acetylsalicylicyl chloride 6,which was treated without further purification with 4a–l in the presence of triethylamine,respectively, and then hydrolyzed with aqueous NaOH to gain intermediates 8a–l. Finally,8a–l were further treated with NH2OK to get N-hydroxycinnamamide/SA hybrids 9a–l,which were purified by column chromatography. The MS data,1H NMR spectra and elemental analysis (EA) of selected compounds are presented in reference [23].

|
Download:
|
| Scheme 1.Reagents and conditions: (a) SOCl2,MeOH,0°C-reflux,8 h,82%; (b) dibromoalkane,K2CO3,CH3CN,reflux,4–6 h,63–77%; (c) ammonium hydroxide or alkylamine, K2CO3,THF,40°C,3–5 h,71–80%; (d) oxalylchloride,CH2Cl2,r.t.,4 h; (e) 4a–l,Et3N,CH2Cl2,r.t.,4–8 h,66–75%; (f) 1 mol/L NaOH (aq.),MeOH,r.t.,2–4 h,85–92%; (g) NH2OK, MeOH,38– 56%. | |
General synthetic procedure for 3a–c: A mixture of methyl ferulicate 2 (2.1 g,10 mmol),K2CO3 (3.5 g,25 mmol) and various dibromoalkanes (30 mmol) in acetonitrile (80 mL) was refluxed for 4–6 h. The solvent was evaporated under vacuum and water was added. The solution was neutralized with 2 mol/L HCl solution to pH 7 ~ 8,and then extracted with CH2Cl2 (60 mL × 3). The organic layer was concentrated in vacuo and the residue was purified by column chromatography on silica gel (petroleum ether–ethyl acetate = 10:1 ~ 6:1,v/v as the eluent) to yield 3a–c as yellowish oil (63–77%).
General synthetic procedure for 4a–l: A mixture of 3a–c (2 mmol),K2CO3 (0.7 g,5 mmol),and excess of ammonium hydroxide or alkylamine (10 mmol) in THF (40 mL) was stirred for 3–5 h at 40°C. The solvent was evaporated under vacuum and water was added. The solution was neutralized to pH 8,and then extracted with ethyl acetate (40 mL × 3). The organic layer was combined,washed with brine,dried with anhydrous Na2SO4,and concentrated in vacuo,affording 4a–l as pale yellow oil (71–80%). General synthetic procedure for 7a–l: Acetylsalicylic acid 5 (1 mmol) was reacted with oxalyl chloride (3 mmol) for 4 h at room temperature. The solvent was evaporated to obtain the salicylicyl chloride 6,which was subsequently added to the mixture of 4a–l (0.9 mmol) and TEA (1.1 mmol) in 30 mL CH2Cl2 at 0°C over 1 h. After stirring for another 0.5 h,the mixture was poured into CH2Cl2. The solution was washed with 5% sodium bicarbonate and brine,dried with anhydrous Na2SO4,and concentrated in vacuo to afford compound 7a–l as pale yellow solid (66–75%).
General synthetic procedure for 8a–l: To a solution of 7a–l (0.5 mmol) in methanol (10 mL),1 mol/L NaOH (1.5 mL) was added drop wise at room temperature. The mixture was refluxed for 2–4 h,and then cooled. The solvent was evaporated and the residue was dissolved in water. The mixture was neutralized with 2 mol/L HCl solution to pH 6 - 7 and extracted with ethyl acetate (30 mL × 3). The organic layers were combined,washed with brine,dried with anhydrous Na2SO4,and concentrated in vacuo, affording 8a–l as pale yellow waxy solid (85–92%). General synthetic procedure for 9a–l: To a solution of compound 8a–l (0.4 mmol) in 5 mL of anhydrous methanol was added a solution of NH2OK (0.09 g,4 mmol) in 3 mL of anhydrous methanol. The mixture was stirred for 0.5 h and the solvent was evaporated under vacuum. The residue was acidified with 2 mol/L HCl to pH 5–6,then extracted with ethyl acetate (10 mL × 3). The organic layers were combined,washed with brine (10 mL × 3), dried over anhydrous Na2SO4,and evaporated with the residue being purified by column chromatography on silica gel (ethyl acetate/methanol,10:1 ~ 6:1,v/v as the eluent),yielding 9a–l as yellowish solid (38–57%). 3. Results and discussion
The inhibitory activities of synthetic compounds 8e and 9a–l against six human cancer cells SGC7901,HCT-15,HCT-116,MCF-7, HeLa,and SKOV-3 were evaluated by MTT assays in vitro,with SAHA used as positive control. The results (shown in Table 1) illustrated the IC50 values of target compounds against each tumor cell line. Compounds 9b–f and 9h showed good to moderate cytotoxic activities. Among these compounds,compound 9e (IC50 = 4.11-7.57 mmol/L) exhibited more potent antiproliferative activities,which were comparable to,or slightly weaker than,SAHA (IC50 = 2.11-7.23 mmol/L). However,the intermediate 8e of 9e displayed poor cancer inhibitory activities,which suggested that the hydroxamic acid moiety has a positive influence on the cytotoxicity of this series of N-hydroxyferulicylamide derivatives.
| Table 1 The IC50 values of 8e and 9a–l against six human cancer cell lines. |
Furthermore,the active compounds 9b–f were evaluated for their in vitro HDAC inhibitory activities against HeLa cell nuclear extract (containing primarily HDAC1 and HDAC2),in addition to HDAC3 and HDAC6. Results,listed in Table 2,showed that 9c–f also exhibited significant HDACs inhibition,which were comparable to SAHA. As expected,the HDAC inhibitory effects of individual compounds were associated with their cytotoxicities against these cancer cells in vitro. In addition,the synthetic compounds 9c–f did not exhibit any isoform selectivity in the HDAC inhibitory assay.
| Table 2 The HDAC inhibitory activities of 9b–f. |

{kind=link}
{kind=link}
Given that most N-hydroxyferulicylamide derivatives exhibit strong cancer and HDAC inhibition,we questioned (or desired to confirm) whether those compounds were toxic to the normal cells. So the cytotoxicities of 9e with the more prominent anticancer activities were further determined by MTT assay against human normal cells CCD-841,and human colon carcinoma cells HCT-116 and HCT-15. As shown in Fig. 2,the treatment with 12 mmol/L of 9e promoted nearly 90% inhibition in HCT-116 and HCT-15 cells, while the same treatment had no significant effect on the survival of CCD-841 cells (only 15% inhibition). Evidentially,compound 9e had strong cytotoxicity selectively to human cancer cells in vitro.

|
Download:
|
| Fig. 2.Cytotoxicity of 9e against human colon carcinoma cells HCT-116,HCT-15, and non-tumor CCD-841 cells. HCT-116,HCT-15,and CCD-841 cells were cultured in media in the presence of indicated concentrations of 9e for 48 h. The cytotoxicity of 9e was determined by MTT assay. Data are means ± SEM of cytotoxicity (%) from three independent experiments. | |
{kind=link}
Analysis of SAR revealed that the carbochain lengths of both the substituted alkyl amides and alkylol amines linking salicylic acid and N-hydroferulicylamide were crucial for their anticancer activity in vitro. Variations in the carbochain lengths of alkylol amines affected significantly the in vitro anticancer activity of these derivatives. For example,the compounds 9b,9e,and 9h with a three-carbon alkylol amine showed relatively stronger anticancer activity than compounds with a two- or four-carbon linker. It is likely that the compounds with three-carbon alkylol amines bind relatively well to the receptor. Among the N-hydroxycinnamamide/ SA hybrids 9b,9e,9h,and 9k with three-carbon alkylol amines,compound 9e displayed the most potent activity against tumor cells,suggesting that adding one atom to the chain length of alkyl amides in N-hydroferulicylamide hybrids may be the optimal modification,and extending or shortening the chain length leads to the decrease of the anticancer activities in vitro. A further investigation of the precise SAR of these compounds is ongoing. 4. Conclusion
In summary,a series of novel HDAC inhibitors 9a–l as Nhydroxycinnamamide/ SA hybrids were designed and synthesized by coupling the carboxyl group of SA with N-hydroxycinnamamides through various alkylol amines. The in vitro assay of their inhibitory activities against six tumor cell lines showed that the Nhydroxycinnamamide/ SA hybrids displayed strong antitumor activities. Especially,compound 9e had greater potency,comparable to SAHA,in human colon carcinoma cells,which was probably, or at least partially,attributable to the three-carbon chain length of alkylol amines. Furthermore,the HDAC inhibitory activities of 9e against HeLa cell nuclear demonstrated more potency than that of SAHA,while compounds 9c–f did not exhibit any isoform selectivity in the HDAC inhibitory assay. In addition,compound 9e could selectively inhibit tumor cells,but not human normal cell proliferation in vitro. Further investigations are ongoing for more precise SAR information and more potent agents.
| [1] | T.K. Kelly, D.D. de Carvalho, P.A. Jones, Epigenetic modifications as therapeutic targets, Nat. Biotechnol. 28 (2010) 1069-1078. |
| [2] | L.P. Blair, Q. Yan, Epigenetic mechanisms in commonly occurring cancers, DNA Cell Biol. 31 (2012) S49-S61. |
| [3] | H. Lehrmann, L.L. Pritchard, A. Harel-Bellan, et al., Histone acetyltransferases and deacetylases in the control of cell proliferation and differentiation, Adv. Cancer. Res. 86 (2002) 41-65. |
| [4] | O. Witt, H.E. Deubzer, T. Milde, I. Oehme, HDAC family: what are the cancer relevant targets, Cancer Lett. 277 (2009) 8-21. |
| [5] | A.A. Lane, B.A. Chabner, Histone deacetylase inhibitors in cancer therapy, J. Clin. Oncol. 27 (2009) 5459-5468. |
| [6] | B.E. Gryder, Q.H. Sodji, A.K. Oyelere, Targeted cancer therapy: giving histone deacetylase inhibitors all they need to succeed, Future Med. Chem. 4 (2012) 505-524. |
| [7] | P.A. Marks, The clinical development of histone deacetylase inhibitors as targeted anticancer drugs, Expert Opin. Invest. Drugs 19 (2010) 1049-1066. |
| [8] | H.S. Wang, B.W. Dymock, New patented histone deacetylase inhibitors, Expert Opin. Ther. Pat. 19 (2009) 1727-1757. |
| [9] | M. Paris, M. Porcelloni, M. Binaschi, D. Fattori, Histone deacetylase inhibitors: from bench to clinic, J. Med. Chem. 51 (2008) 1505-1529. |
| [10] | P.A. Marks, Discovery and development of SAHA as an anticancer agent, Oncogene 26 (2007) 1351-1356. |
| [11] | C. Campas-Moya, Romidepsin for the treatment of cutaneous T-cell lymphoma, Drugs Today 45 (2009) 787-795. |
| [12] | P.A. Cassier, A. Lefranc, E.Y. Amela, et al., A phase Ⅱ trial of panobinostat in patients with advanced pretreated soft tissue sarcoma: a study from the French Sarcoma Group, |
| [13] | J.A. Plumb, P.W. Finn, R.J. Williams, et al., Pharmacodynamic response and inhibition of growth of human tumor xenografts by the novel histone deacetylase inhibitor PXD101, Mol. Cancer Ther. 2 (2003) 721-728. |
| [14] | A.R. Razak, S.J. Hotte, L.L. Siu, et al., Phase I clinical, pharmacokinetic and pharmacodynamic study of SB939, an oral histone deacetylase (HDAC) inhibitor, in patients with advanced solid tumours, |
| [15] | C. Doñas, M. Fritz, V. Manríquez, et al., Trichostatin A promotes the generation and suppressive functions of regulatory T cells, Clin. Dev. Immunol. (2013) 679-804. |
| [16] | K. Rao-Bindal, N.V. Koshkina, J. Stewart, et al., The histone deacetylase inhibitor, MS-275 (entinostat), downregulates c-FLIP, sensitizes osteosarcoma cells to FasL, and induces the regression of osteosarcoma lung metastases, Curr. Cancer Drug Targets 13 (2013) 411-422. |
| [17] | Y. Boumber, A. Younes, G. Garcia-Manero, Mocetinostat (MGCD0103): a review of an isotype-specific histone deacetylase inhibitor, Expert Opin. Investig. Drugs 20 (2011) 823-829. |
| [18] | T.A. Miller, D.J. Witter, S. Belvedere, Histone deacetylase inhibitors, J. Med. Chem. 46 (2003) 5097-5116. |
| [19] | Z.H. Zhao, M.H. Moghadasian, Chemistry, natural sources, dietary intake and pharmacokinetic properties of ferulic acid: a review, Food Chem. 109 (2008) 691-702. |
| [20] | M.D. Lu, X. Zhou, Y.J. Yu, et al., Synthesis and in vitro biological evaluation of nitric oxide-releasing derivatives of hydroxylcinnamic acids as anti-tumor agents, Chin. Chem. Lett. 24 (2013) 415-418. |
| [21] | A. Ghasemzadeh, H.Z. Jaafar, E. Karimi, Involvement of salicylic acid on antioxidant and anticancer properties, anthocyanin production and chalcone synthase activity in ginger (Zingiber officinale Roscoe) varieties, Int. J. Mol. Sci. 13 (2012) 14828-14844. |
| [22] | J. Sonnemann, I. Hü ls, M. Sigler, et al., Histone deacetylase inhibitors and aspirin interact synergistically to induce cell death in ovarian cancer cells, Oncol. Rep. 20 (2008) 219-224. |
| [23] | The data of selected compounds. 9b: Yield 46%, mp 111-114 ℃; MS (ESI): m/z 387[M + H]+; 1H NMR (300 MHz, DMSO-d6): δ 10.45 (s, 1H), 8.96 (s, 1H), 7.66-7.78 (m, 2H), 7.42 (d, 1H, J = 16.2 Hz), 6.78-7.12 (m, 5H), 6.38 (d, 1H, J = 16.2 Hz), 4.12 (t, 2H, J = 4.5 Hz), 3.83 (s, 3H), 3.62 (t, 2H, J = 4.5 Hz), 1.96-2.21 (m, 2H); Anal. Calcd. for C20H22N2O6: C, 62.17; H, 5.74; N, 7.25; Found: C, 62.28; H, 5.56; N, 7.13. 9c: Yield 52%, mp 102-104 ℃; MS (ESI): m/z 401 [M + H]+; 1H NMR (300 MHz, DMSO-d6): δ 10.41 (s, 1H), 8.89 (s, 1H), 7.65-7.77 (m, 2H), 7.40 (d, 1H, J = 16.2 Hz), 6.73-7.15 (m, 5H), 6.35 (d, 1H, J = 16.2 Hz), 4.10 (t, 2H, J = 4.5 Hz), 3.80 (s, 3H), 3.59 (t, 2H, J = 4.5 Hz), 1.75-1.98 (m, 4H); Anal. Calcd. for C21H24N2O6: C, 62.99; H, 6.04; N, 7.00; Found: C, 62.85; H, 4.96; N, 7.13. 9d: Yield 38%, mp 119-122 ℃; MS (ESI): m/z 387 [M + H]+; 1H NMR (300 MHz, DMSO-d6): δ 10.44 (s, 1H), 7.68-7.79 (m, 2H), 7.45 (d, 1H, J = 16.2 Hz), 6.79-7.21 (m, 5H), 6.39 (d, 1H, J = 16.2 Hz), 4.15 (t, 2H, J = 4.5 Hz), 3.84 (s, 3H), 3.65 (t, 2H, J = 4.5 Hz), 3.06 (s, 3H); Anal. Calcd. for C20H22N2O6: C, 62.17; H, 5.74; N, 7.25; Found: C, 62.03; H, 5.86; N, 7.17. 9e: Yield 47%, mp 108-111 ℃; MS (ESI): m/z 401 [M + H]+; 1H NMR (300 MHz, DMSO-d6): δ 10.42 (s, 1H), 7.65-7.78 (m, 2H), 7.43 (d, 1H, J = 16.2 Hz), 6.76-7.19 (m, 5H), 6.37 (d, 1H, J = 16.2 Hz), 4.13 (t, 2H, J = 4.5 Hz), 3.81 (s, 3H), 3.64 (t, 2H, J = 4.5 Hz), 3.03 (s, 3H), 1.98-2.23 (m, 2H); Anal. Calcd. for C21H24N2O6: C, 62.99; H, 6.04; N, 7.00; Found: C, 63.07; H, 6.26; N, 7.16. 9f: Yield 55%, mp 103-106 ℃; MS (ESI): m/z 415[M + H]+; 1H NMR (300 MHz, DMSO-d6): δ 10.47 (s, 1H), 7.63-7.77 (m, 2H), 7.40 (d, 1H, J = 16.2 Hz), 6.73-7.17 (m, 5H), 6.34 (d, 1H, J = 16.2 Hz), 4.10 (t, 2H, J = 4.5 Hz), 3.79 (s, 3H), 3.61 (t, 2H, J = 4.5 Hz), 3.02 (s, 3H), 1.78-2.02 (m, 4H); Anal. Calcd. for C22H26N2O6: C, 63.76; H, 6.32; N, 6.76; Found: C, 63.57; H, 6.56; N, 7.59. 9h: Yield 57%, mp 95-98 ℃; MS (ESI): m/z 415 [M + H]+; 1H NMR (300 MHz, DMSO-d6): δ 10.39 (s, 1H), 7.59-7.75 (m, 2H), 7.38 (d, 1H, J = 16.2 Hz), 6.70-7.16 (m, 5H), 6.32 (d, 1H, J = 16.2 Hz), 4.07 (t, 2H, J = 4.5 Hz), 3.80 (s, 3H), 3.57 (t, 2H, J = 4.5 Hz), 3.00 (s, 3H), 1.95-2.22 (m, 2H); Anal. Calcd. for C22H26N2O6: C, 63.76; H, 6.32; N, 6.76; Found: C, 63.53; H, 6.48; N, 7.62. |