




Dihydropyrimidinones (DHPMs) and their derivatives have recently attracted great attention in synthetic organic chemistry due to their pharmacological and therapeutic properties,such as antibacterial and antihypertensive activity as well as behaving as calcium channel blockers,a-1a-antagonists [1] and neuropeptide Y (NPY) antagonists [2]. The biological activity of some alkaloids isolated recently has been attributed to a dihydropyrimidinone moiety [3]. The first synthesis of these compounds,reported by Biginelli [4] more than a century ago,makes use of the three components,one-pot condensation of a &deta;-ketoester,an aldehyde and urea under strongly acidic conditions [5]. However,this method suffers from low yields in the case of substituted aromatic and aliphatic aldehydes [6]. Owing to the versatile biological activity of dihydropyrimidinones,development of an alternate synthetic methodology is of paramount importance.
In recent years,several synthetic procedures for the preparation of DHPMs have been reported including classical conditions with microwave irradiation [7] and by using Lewis acids,as well as protic acids and silica supported solid acids as promoters. Such promoters as conc. HCl [8],BF3.OEt2 [9],PPE [10],KSF clay [11], InCl3 [12],LaCl3 [13, 14],lanthanide triflate [15],H2SO4 [16],ceric ammonium nitrate (CAN) [17],Mn(OAc)3 [18],ion-exchange resin [19],1-n-butyl-3-methyl imidazolium tetrafluoroborate (BMImBF4) [20],BiCl3 [21],LiClO4 [22],InBr3 [23],FeCl3 [24], ZrCl4 [25],Cu(OTf)2 [26],Bi(OTf)3 [27],Silica gel-supported Lpyrrolidine- 2-carboxylic acid-4-hydrogen sulfate [28],SBA-15 sulfonic acid [29],silica sulfuric acid [30],PEG–SO3 [31],pdodecylbenzenesulfonic acid (DBSA) [32],etc. have been found to be effective. Many of these methods involve expensive reagents, stoichiometric amounts of catalysts,strongly acidic conditions, long reaction times,unsatisfactory yields and incompatibility with other functional groups. Therefore,the development of a neutral alternative would extend the scope of the Biginelli reaction. The heterogeneous catalysts,useful in making the organic transformations eco-friendly and economically viable,are now in high demand in academic laboratories and industries. Several heterogeneous catalysts are thus finding increasing applications in the field of catalysis [33]. Solid acid-based catalysts have offered simpler,more reactive and more benign alternatives than their homogeneous counterparts [34]. The expensive,organic polymer chain in traditional polymer supported catalysts has now been replaced by a silica chain having a covalently anchored organic spacer to create organic-inorganic hybrid (interphase) catalysts [35]. In these heterogeneous catalysts,the reactive centers are highly mobile similar to homogeneous catalysts and at the same time they can be recovered. Based on these concepts,various types of functionalized,sulfonic acid silica,as Bronsted acid sites,have been considered in selectively catalyzing chemical transformations can be created [35, 36].
The utility of heterogeneous catalysts can be improved by using high surface area materials,since the transport of the reactants to the active site on the particle surface can be enhanced by this method. Mesoporous silicas functionalized with sulfonic acid groups have been obtained by the condensation of alkoxy silanes and 3-mercapto propyl trimethoxy silanes,which are further oxidized to obtain their corresponding propyl sulfonic acid groups [37]. Shi et al. [38] had recently reported on a sulfonic acidfunctionalized polypropylene fiber catalyst which was prepared by a simple process from inexpensive,readily available polypropylene fiber and the performance of the catalyst was tested in the successful completion of the Biginelli reaction. Excellent results,in terms of chemical yields,catalyst levels,work-up procedures and catalytic recyclability,are reported on the metal-free catalyzed, one-pot synthesis of a number of substituted 3,4-dihydropyrimidin- 2-(1H)-ones/-thiones (DHPMs).
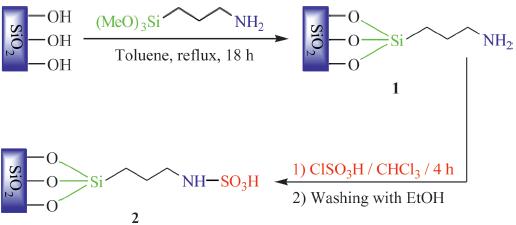
In this report,we describe a one-pot method for the Biginelli reaction using silica-bonded N-propyl sulfamic acid (SBNPSA) as a recyclable,heterogeneous,solid acid catalyst. The reaction involves the synthesis of a variety of 3,4-dihydropyrimidin- 2(1H)-ones by the reaction of b-ketoester with urea (or thiourea) and various aromatic aldehydes in ethanol at reflux temperatures. The preparation procedure for catalyst 2 is outlined in Scheme 1 with slight modification than the previously reported method [35].

|
Download:
|
| Scheme 1.Preparation of silica-bonded N-propyl sulfamic acid (2). | |
Reagents and solvents were of analytical grade or were purified by standard procedures prior to use. The 1H NMR spectra were recorded on Varian FT-200 MHz (Gemini) in DMSO-d6. Chemicals shifts are reported in parts per million (δ) relative to tetramethylsilane (δ 0.0) as an internal standard. Mass spectra were recorded under electron impact at 70 eV on Finnigan Mat 1020B mass spectrometer. Melting points were recorded on Buchi 535 and are uncorrected. Thin-layer chromatography was performed on Merck 60 F-254 silicagel gel plates. Products obtained are all known compounds and were identified by comparing their physical and spectral data with those reported in the literature. 2.2. Catalyst preparation
To a mixture of 3-aminopropylsilica 1 (5 g) in chloroform (20 mL),chlorosulfonic acid (1 g,0.6 mL) was added dropwise at 0 8C over 2 h. After addition was complete,the mixture was stirred for 2 h until HCl gas evolution stopped. Then,the mixture was filtered and washed with ethanol (30 mL) and dried at room temperature to obtain silica-bonded N-propyl sulfamic acid (2) as a cream colored powder (5.13 g). The nitrogen content of 3- aminopropylsilica (1) and sulfur content of silica-bonded N-propyl sulfamic acid (2) determined by conventional elemental analysis were 5.13% and 9.29%,respectively. 2.3. General procedure for the synthesis of 3,4-dihydropyrimidin- 2(1H)-ones and -thiones 4(a–n) using SBNPSA as catalyst
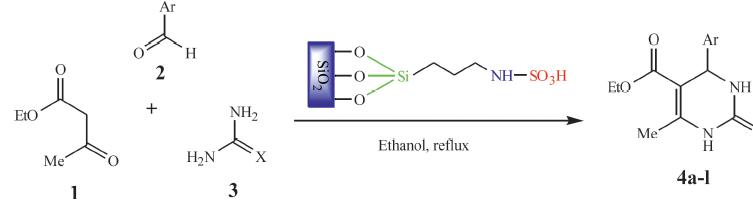
To a mixture of ethyl acetoacetate 1 (2.5 mmol),aromatic aldehyde 2 (2.5 mmol) and urea or thiourea 3 (2.5 mmol) in a round bottomed flask (100 mL),ethanol (15 mL) was added (Scheme 2). The reaction mixture was stirred at r.t. for 5 min and then catalyst 2 (0.2 g,2.4 mol% of SO3H) was added and the stirring was continued at 80 8C for an appropriate time. After completion of the reaction (monitored by TLC),the reaction mixture was filtered off. The product was obtained after removal of the solvent under reduced pressure followed by treatment with water and finally crystallization from EtOH. The residue was washed with warm ethanol,dried at 100°C for 2 h and re-used for eight times without loss of significant activity.

|
Download:
|
| Scheme 2.SBNPSA catalyzed synthesis of 3,4-dihydropyrimidin-2-(1H)-ones/thiones. | |
5-(Ethoxycarbonyl)-6-methyl-4-phenyl-3,4-dihydropyrimidin- 2(1H)-one (4a): 1H NMR (DMSO-d6): δ 1.09 (t,3H,J = 7.1 Hz, OCH2CH3),2.25 (s,3H,CH3),3.97 (q,2H,J = 7.1Hz,OCH2),5.05 (d,1H,J = 2.15 Hz,–CH),7.28 (m,5H,Ar–H),7.75 (s,1H,NH),9.20 (s,1H,NH); 13C NMR (DMSO-d6): δ 14.11,17.94,54.91,60.05, 100.95,112.85,113.05,125.15,125.81,129.05,131.20,150.16, 155.47,163.81; IR (KBr,cm-1): νmax 3240,1722,1638; ESI-MS 261 (M+ H); HRMS calcd. for C1H16N2O3: 260.1161,found: 260.1163.
5-(Ethoxycarbonyl)-4-(4-nitrophenyl)-6-methyl-3,4-dihydropyrimidin- 2(1H)-one (4b): 1H NMR (DMSO-d6): δ 1.11 (t,3H,J = 7.04 Hz,OCH2CH3),2.32 (s,3H,CH3),4.03 (q,2H,J = 7.12 Hz, OCH2CH3),5.78 (d,1H,J = 2.28 Hz,–CH),7.51 (d,2H,J = 9.18 Hz, Ar–H),7.69 (s,1H,NH),8.16 (d,2H,J = 9.16 Hz,Ar–H),9.05 (s,1H,NH); 13C NMR (DMSO-d6): δ 14.22,18.71,55.81,60.15, 101.60,118.15,130.37,138.34,152.26,153.41,159.15,165.85; IR (KBr,cm-1): νmax 3235,1740,1631; ESI-MS 306 (M + H); HRMS calcd. for C14H15N3O5: 305.1012,found: 305.1010.
5-(Ethoxycarbonyl)-4-(3-chlorophenyl)-6-methyl-3,4-dihydropyrimidin- 2(1H)-one (4c): 1H NMR (DMSO-d6): δ 1.10 (t,3H,J = 7.14 Hz,OCH2CH3),2.28 (s,3H,CH3),3.88 (q,2H,J = 7.16 Hz, OCH2CH3),5.65 (d,1H,J = 2.28 Hz,–CH),7.25–7.41 (m,4H, Ar–H),7.61 (s,1H,NH),9.11 (s,1H,NH); 13C NMR (DMSO-d6): δ 14.17,18.60,55.70,60.20,101.52,126.312,127.92,128.42,130.29, 135.51,142.21,153.23,159.32,165.75; IR (KBr,cm-1): νmax 3234, 1724,1631; ESI-MS 295 (M + H); HRMS calcd. for C14H15ClN2O3: 294.0771,found: 294.0772.
5-(Ethoxycarbonyl)-4-(4-chlorophenyl)-6-methyl-3,4-dihydropyrimidin- 2(1H)-one (4d): 1HNMR (DMSO-d6): δ 1.12 (t,3H, J = 7.14 Hz,OCH2CH3),2.30 (s,3H,CH3),3.91 (q,2H,J = 7.16 Hz, OCH2CH3),5.70 (d,1H,J = 2.28,–CH),7.21 (d,2H,J = 9.18,Ar–H), 7.69 (s,1H,NH),7.94 (d,2H,J = 9.18,Ar–H),9.16 (s,1H,NH); 13C NMR (DMSO-d6) d: 14.18,18.62,55.72,60.21,101.55,118.17, 130.32,142.29,152.31,153.39,159.17,165.83; IR (KBr,cm-1): νmax 3225,1720,1615; ESI-MS 295 (M+H); HRMS calcd. for C14H15ClN2O3: 294.0771,found:294.0773.
5-(Ethoxycarbonyl)-4-(3-bromophenyl)-6-methyl-3,4-dihydropyrimidin- 2(1H)-one (4e): 1H NMR (DMSO-d6): δ 1.02 (t,3H, J = 7.05 Hz,OCH2CH3),2.30 (s,3H,CH3),3.75 (q,2H,J = 7.05 Hz, OCH2CH3),5.41 (d,1H,J = 2.25 Hz,–CH),7.05–7.34 (m,4H,Ar–H), 7.51 (s,1H,NH),9.05 (s,1H,NH); 13C NMR (DMSO-d6): δ 14.16, 18.59,55.74,60.18,101.57,126.35,127.82,128.48,130.32,135.59, 143.94,153.21,159.30,165.74; IR (KBr,cm-1): νmax 3212,1731, 1620; ESI-MS 339 (M+H); HRMS calcd. for C14H15BrN2O3: 338.0266,found: 338.0268.
5-(Ethoxycarbonyl)-4-(4-methoxyphenyl)-6-methyl-3,4-dihydropyrimidin- 2(1H)-one (4f): 1H NMR (DMSO-d6) δ: 1.15 (t,3H, J = 7.12 Hz,OCH2CH3),2.33 (s,3H,CH3),3.78 (s,3H,–OCH3),4.06 (q,2H,J = 7.12 Hz,OCH2CH3),5.34 (d,1H,J = 2.28 –CH),6.82 (d,2H, J = 8.60,Ar–H),7.22 (d,2H,J = 8.60,Ar–H),7.76 (s,1H,NH),9.26 (s, 1H,NH); 13C NMR (DMSO-d6) δ:14.32,18.80,55.23,55.40,60.17, 101.68,114.06,127.97,136.22,146.16,153.59,159.30,165.87; IR (KBr,cm-1): νmax 3232,1720,1638; ESI-MS 291 (M+H); HRMS calcd. for C15H18N2O4: 290.1267,found: 290.1265.
5-(Ethoxycarbonyl)-4-(2,4-dichlorophenyl)-6-methyl-3,4- dihydropyrimidin-2(1H)-one (4g): 1H NMR (DMSO-d6): δ 1.18 (t,3H,J = 7.23 Hz,OCH2CH3),2.64 (s,3H,CH3),4.07 (q,2H, J = 7.24 Hz,OCH2CH3),5.92 (d,1H,J = 2.30 Hz,–CH),7.21–7.51 (m, 3H,Ar–H),7.69 (s,1H,NH),9.16 (s,1H,NH); 13C NMR (DMSO-d6): δ 14.20,18.60,55.75,60.24,101.56,127.82,128.91,129.52,131.29, 142.52,143.25,153.23,159.32,165.75; IR (KBr,cm-1): νmax 3255, 1731,1651; ESI-MS 329 (M+H); HRMS calcd. for C14H14Cl2N2O3: 328.0381,found: 328.0379.
5-(Ethoxycarbonyl)-4-(3-nitrophenyl)-6-methyl-3,4-dihydropyrimidin- 2(1H)-one (4 h): 1H NMR (DMSO-d6): δ 1.12 (t, 3H,J = 7.10 Hz,OCH2CH3),2.25 (s,3H,CH3),3.65 (q,2H, J = 7.14 Hz,OCH2CH3),5.71 (d,1H,J = 2.20 Hz,–CH),7.21–7.54 (m,4H,Ar–H),7.74 (s,1H,NH),9.26 (s,1H,NH); 13C NMR (DMSOd6): δ 14.16,18.59,55.74,60.18,101.57,126.25,127.45,128.74, 130.56,135.46,144.81,153.64,159.45,165.30; IR (KBr,cm-1): νmax 3229,1724,1630; ESI-MS 306 (M+H); HRMS calcd. for C14H15N3O5: 305.1012,found: 305.1013.
5-(Ethoxycarbonyl)-4-(4-flurophenyl)-6-methyl-3,4-dihydropyrimidin- 2(1H)-one (4i): 1H NMR (DMSO-d6): δ 1.15 (t,3H, J = 7.16 Hz,OCH2CH3),2.41 (s,3H,CH3),4.12 (q,2H,J = 7.17 Hz, OCH2CH3),5.88 (d,1H,J = 2.25 Hz,–CH),7.69 (s,1H,NH),7.81 (d,2H,J = 8.5 Hz,Ar–H),7.94 (d,2H,J = 9.18 Hz,Ar–H),9.16 (s,1H, NH); 13C NMR (DMSO-d6): δ 14.18,18.62,55.72,60.21,101.55, 121.19,132.42,144.20,153.39,157.25,159.17,165.83; IR (KBr, cm-1): νmax 3250,1741,1654; ESIMS 279 (M+H); HRMS calcd. for C14H15N2O5: 278.1067,found: 278.1069.
5-(Ethoxycarbonyl)-6-methyl-4-phenyl-3,4-dihydropyrimidin- 2(1H)-thione (4j): 1H NMR (DMSO-d6): δ 1.11 (t,3H, J = 7.21 Hz,OCH2CH3),2.29 (s,3H,CH3),4.12 (q,2H, J = 7.24 Hz,OCH2),5.16 (d,1H,J = 2.05 Hz,–CH),7.51 (m,5H, Ar–H),7.81 (s,1H,NH),9.41 (s,1H,NH); 13C NMR (DMSO-d6): δ 14.23,17.91,54.85,60.15,100.90,112.84,115.12,125.15, 126.85,129.64,131.45,150.27,162.63,180.25; IR (KBr,cm-1): νmax 3240,1720,1640,1595,1530; ESI-MS 277 (M+H); HRMS calcd. for C14H16N2O2S: 276.0932 found: 276.0932.
5-(Ethoxycarbonyl)-4-(4-methoxyphenyl)-6-methyl-3,4-dihydropyrimidin- 2(1H)-thione (4k): 1H NMR (DMSO-d6): δ 1.17 (t, 3H,J = 7.11 Hz,OCH2CH3),2.37 (s,3H,CH3),4.12 (s,3H,–OCH3), 4.15 (q,2H,J = 7.10 Hz,OCH2CH3),5.44 (d,1H,J = 2.15 Hz,–CH), 7.11 (d,2H,J = 8.15 Hz,Ar–H),7.37 (d,2H,J = 8.11 Hz,Ar–H),7.84 (s,1H,NH),9.43 (s,1H,NH); 13C NMR (DMSO-d6): δ 14.32,18.05, 55.24,55.49,60.45,101.84,114.32,127.74,137.25,147.15,159.45, 165.62,182.48; IR (KBr,cm-1): νmax 3240,1725,1635,1574,1540; ESI-MS 307 (M+H); HRMS calcd. for C15H18N2O3S: 306.1038, found: 306.1040.
5-(Ethoxycarbonyl)-4-(3-nitrophenyl)-6-methyl-3,4-dihydropyrimidin- 2(1H)-thione (4l): 1H NMR (DMSO-d6): δ 1.15 (t,3H, J = 7.14 Hz,OCH2CH3),2.27 (s,3H,CH3),4.02 (q,2H,J = 7.11 Hz, OCH2CH3),5.81 (d,1H,J = 2.06 Hz,–CH),7.23–7.37 (m,4H,Ar–H), 7.78 (s,1H,NH),9.34 (s,1H,NH); 13C NMR (DMSO-d6): δ 14.14, 18.60,55.64,60.21,101.34,126.25,128.02,129.32,130.75, 135.65,144.34,160.40,165.64,182.65; IR (KBr,cm-1): νmax 3245,1725,1632,1575,1545; ESI-MS 322 (M+H); HRMS calcd. for C14H15N3O4S: 321.0783,found: 321.0781. 3. Results and discussion
In the FT-IR spectrum of catalyst (2),the major peaks for silica (SiO2) are broad non-symmetric Si–O–Si stretching from 1300 cm-1 to 1010.6 cm-1 and symmetric Si–O–Si stretching near 880–852.5 cm-1. For sulfuric acid functional group,the FT-IR absorption range of the O–S–O asymmetric and symmetric stretching modes lie in 1170 anδ 1060 cm-1,respectively. FT-IR spectrum shows the overlap asymmetric and symmetric stretching bands of SO2 with Si–O–Si stretching bands in the silica functionalized alkyl-sulfuric acid. The spectrum also shows a broad OH stretching absorption around 3600–2520 cm-1.
The BET surface area and total pore volume of Silica-bonded Npropylsulfamic acid (2) were found to be 2.91 m2 g-1 and 0.456 cm3 g-1,respectively.
In order to optimize the reaction conditions,the synthesis of compound 4f was used as a model reaction. Therefore,a mixture of ethyl acetoacetate (2.5 mmol),4-methoxybenzaldehyde (2.5 mmol),and urea (2.5 mmol) with different amounts of SBNPSA (Table 1) was selected. The efficiency of the reaction is mainly affected by the amount of the catalyst. No product could be detected in the absence of this catalyst even after 12 h (entry 1), while good results were obtained in the presence of SBNPSA. The optimal amount of the catalyst was 0.2 g (entry 5),whereas a higher amount of the catalyst did not increase the yield noticeably (entry 6).
| Table 1 Influence of the amount of SBNPSA on the synthesis of 4f at reflux temperature.a |

As can be seen from (Table 2) compared to the classical Biginelli method,one additional important feature of the present protocol is the ability to tolerate variations in all three components simultaneously. Most importantly,many of the pharmacological relevant substitution patterns on the aromatic ring could be introduced without any reduction in efficiency. Aromatic aldehydes carrying either electron-donating or electron-withdrawing substituents afforded high yields of products in high purity. Thiourea has been used with similar success to provide the corresponding dihydropyrimidin-2(1H)-thiones (4j–4l),which are also of much interest with regard to biological activity. Thus, variation in all three components has been accomplished conveniently.
| Table 2 SBNPSA catalyzed synthesis of 3,4-dihydropyrimidin-2-(1H)-ones and thiones. |

Table 3 summarizes our data (time,yield,reaction conditions) with results obtained by other groups. Based on this comparison, our method is simpler,more efficient for the synthesis of dihydropyrimidinone derivatives.
| Table 3 Comparison our results with results obtained by other groups. |

{kind=link}
{kind=link}
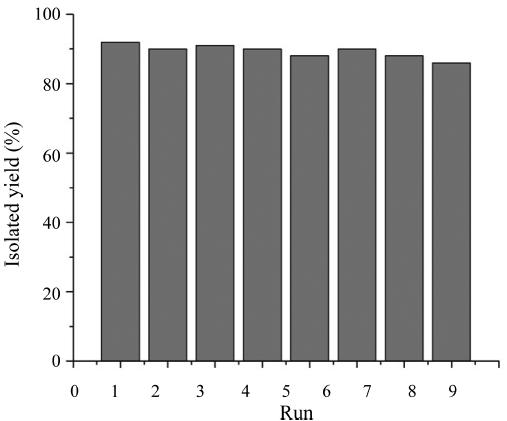
The recycling of the catalyst was also investigated. For this purpose,the same model reaction to synthesize the compound 4f was again studied under the optimized conditions. After completion of the reaction,the catalyst was filtered,washed with warm ethanol,dried at 100°C under vacuum for 2 h and reused for the same reaction process. As shown in Fig. 1,the catalyst could be reused for eight times with only slight reduction in the catalytic activity of the catalyst.

|
Download:
|
| Fig. 1.Recycling of SBNPSA for model reaction. | |
{kind=link}
We have developed a simple,efficient,and green protocol for the synthesis of dihydropyrimidinones using silica-bonded Npropyl sulfamic acid catalyst under heterogeneous conditions. The short reaction times,simple work-up in the isolation of the products in high yields with high purity,mild reaction conditions, and recycability of supported catalyst are noteworthy features of this new procedure. Acknowledgments
The authors are thankful to Madhya Pradesh Council of Science & Technology (MPCOST,Bhopal) for their financial support. Authors are also thankful to Deputy Director and Head,SAIF, Central Drug Research Institute (CDRI),Lucknow,for providing elemental analysis and spectral data and the Department of Chemistry,Vikram University,Ujjain,for extending laboratory facilities and IR data.
| [1] | C.O. Kappe, 100 years of the Biginelli dihydropyrimidine synthesis, Tetrahedron 49 (32) (1993) 6937-6963. |
| [2] | K.S. Atwal, B.N. Swanson, S.E. Unger, et al., Dihydropyrimidine calcium channel blockers. 3.3-Carbamoyl-4-aryl-1,2,3,4-tetrahydro-6-methyl-5-pyrimidinecarboxylic acid esters as orally effective antihypertensive agents, J. Med. Chem. 34 (1991) 806-811. |
| [3] | B.B. Snider, Z.P. Shi, Biomimetic synthesis of (±)-crambines A, B, C1, and C2. Revision of the structure of crambines B and C1, J. Org. Chem. 58 (1993) 3828-3839. |
| [4] | B.B. Snider, J. Chen, A.D. Patil, A. Freyer, Synthesis of the tricyclic portions of batzelladines A, B and D. Revision of the stereochemistry of batzelladines A and D, Tetrahedron Lett. 37 (1996) 6977-6980. |
| [5] | C.O. Kappe, Recent advances in Biginelli dihydropyrimidine synthesis. New tricks from an old dog, Acc. Chem. Res. 33 (2000) 879-888. |
| [6] | K. Folkers, H.J. Harwood, T.B. Johnson, Research on pyrimidines. cxxx. Synthesis of 2-keto-1,2,3,4-tetrahydropyrimidines, J. Am. Chem. Soc. 54 (1932) 3751-3758. |
| [7] | J.S. Yadav, B.V. Subba Reddy, E. Jagan Reddy, T. Ramalingam, Microwave-assisted efficient synthesis of dihydro pyrimidines: improved high yielding protocol for the Biginelli reaction, J. Chem. Res. Sci. 7 (2000) 354-355. |
| [8] | K.S. Atwal, B.C. O'Reilly, J.Z. Gougoutas, M.F. Malley, Synthesis of substituted 1,2,3,4-tetrahydro-6-methyl-2-thioxo-5-pyrimidinecarboxylic acid esters, Heterocycles 26 (1987) 1189-1192. |
| [9] | E.H. Hu, D.R. Sidler, U.H. Dolling, Unprecedented catalytic three component onepot condensation reaction: an efficient synthesis of 5-alkoxycarbonyl-4-aryl-3,4-dihydro pyrimidin-2(1H)-ones, J. Org. Chem. 63 (1998) 3454-3457. |
| [10] | C.O. Kappe, S.F. Falsone, Polyphosphate ester-mediated synthesis of dihydropyrimidines. Improved conditions for the Biginelli reaction, Synlett 7 (1998) 718-720. |
| [11] | C.O. Kappe, D. Kumar, R.S. Varma, Microwave-assisted high-speed parallel synthesis of 4-aryl-3,4-dihydropyrimidin-2(1H)-ones using a solventless Biginelli condensation protocol, Synthesis 10 (1999) 1799-1803. |
| [12] | F. Bigi, S. Carloni, B. Frullanti, R. Maggi, G. Sartori, A revision of the Biginelli reaction under solid acid catalysis. Solvent-free synthesis of dihydropyrimidines over montmorillonite KSF, Tetrahedron Lett. 40 (1999) 3465-3468. |
| [13] | B.C. Ranu, A. Hajra, U. Jana, Indium(Ⅲ) chloride-catalyzed one-pot synthesis of dihydropyrimidinones by a three-component coupling of 1,3-dicarbonyl compounds, aldehydes, and urea: an improved procedure for the Biginelli reaction, J. Org. Chem. 65 (2000) 6270-6272. |
| [14] | J. Lu, H. Ma, Iron(Ⅲ)-catalyzed synthesis of dihydropyrimidinones. Improved conditions for the Biginelli reaction, Synlett 1 (2000) 63-64. |
| [15] | J. Lu, Y.J. Bai, Z.J. Wang, B.Q. Yang, H.R. Ma, One-pot synthesis of 3,4-dihydropyrimidin-2(1H)-ones using lanthanum chloride as a catalyst, Tetrahedron Lett. 41 (2000) 9075-9078. |
| [16] | Y. Ma, C. Qian, L. Wang, M. Yang, Lanthanide triflate catalyzed Biginelli reaction. One-pot synthesis of dihydropyrimidinones under solvent-free conditions, J. Org. Chem. 65 (2000) 3864-3868. |
| [17] | J.C. Bussolari, P.A. McDonnell, A new substrate for the Biginelli cyclocondensation: direct preparation of 5-unsubstituted 3,4-dihydropyrimidin-2(1H)-ones from a β-keto carboxylic acid, J. Org. Chem. 65 (2000) 6777-6779. |
| [18] | J.S. Yadav, B.V.S. Reddy, K.B. Reddy, K.S. Raj, A.R. Prasad, Ultrasound-accelerated synthesis of 3,4-dihydropyrimidin-2(1H)-ones with ceric ammonium nitrate, J. Chem. Soc., Perkin Trans. 1 (2001) 1939-1941. |
| [19] | K.A. Kumar, M. Kasthuraiah, C.S. Reddy, C.D. Reddy, Mn(OAc)3·2H2O-mediated three-component, one-pot, condensation reaction: an efficient synthesis of 4-aryl-substituted 3,4-dihydropyrimidin-2-ones, Tetrahedron Lett. 42 (2001) 7873-7875. |
| [20] | A. Dondoni, A. Massi, Parallel synthesis of dihydropyrimidinones using Yb(Ⅲ)-resin and polymer-supported scavengers under solvent-free conditions. A green chemistry approach to the Biginelli reaction, Tetrahedron Lett. 42 (2001) 7975-7978. |
| [21] | J. Peng, Y. Deng, Synthesis and revision of the relative configuration of fudecalone, Tetrahedron Lett. 42 (2001) 917-919. |
| [22] | K. Ramalinga, P. Vijayalakshmi, T.N.B. Kaimal, Bismuth(Ⅲ)-catalyzed synthesis of dihydropyrimidinones: improved protocol conditions for the Biginelli reaction, Synlett 6 (2001) 863-865. |
| [23] | J.S. Yadav, B.V. Subba Reddy, R. Srinivas, C. Venugopal, T. Ramalingam, LiClO4-catalyzed one-pot synthesis of dihydropyrimidinones: an improved protocol for Biginelli reaction, Synthesis 9 (2001) 1341-1345. |
| [24] | N.Y. Fu, Y.F. Yuan, Z. Cao, et al., Indium(Ⅲ) |
| [25] | J. Lu, Y.J. Bai, Catalysis of the Biginelli reaction by ferric and nickel chloride hexahydrates. One-pot synthesis of 3,4-dihydropyrimidin-2(1H)-ones, Synthesis 4 (2002) 466-470. |
| [26] | Ch.V. Reddy, M. Mahesh, P.V.K. Raju, T.R. Babu, V.V.N. Reddy, Zirconium(IV) chloride catalyzed one-pot synthesis of 3,4-dihydropyrimidin-2(1H)-ones, Tetrahedron Lett. 43 (2002) 2657-2659. |
| [27] | A.S. Prabhakar, G.K. Dewkar, A. Sudalai, Cu(OTf)2: a reusable catalyst for highyield synthesis of 3,4-dihydropyrimidin-2(1H)-ones, Tetrahedron Lett. 44 (2003) 3305-3308. |
| [28] | A.G. Choghamarani, P. Zamani, Three component reactions: an efficient and green synthesis of 3,4-dihydropyrimidin-2-(1H)-ones and thiones using silica gel-supported L-pyrrolidine-2-carboxylic acid-4-hydrogen sulfate, Chin. Chem. Lett. 24 (2013) 804-808. |
| [29] | S. Rostamnia, F. Pourhassan, The SBA-15/SO3H nanoreactor as a highly efficient and reusable catalyst for diketene-based, four-component synthesis of polyhydroquinolines and dihydropyridines under neat conditions, Chin. Chem. Lett. 24 (2013) 401-403. |
| [30] | S. Rostamnia, K. Lamei, Diketene-based neat four-component synthesis of the dihydropyrimidinones and dihydropyridine backbones using silica sulfuric acid (SSA), Chin. Chem. Lett. 23 (2012) 930-932. |
| [31] | X.C. Wang, L.J. Zhang, Z. Zhang, Z.J. Quan, PEG-OSO3Has an efficient and recyclable catalyst for the synthesis of β-amino carbonyl compounds via the Mannich reaction in PEG-H2O, Chem. Lett. 23 (2012) 423-426. |
| [32] | M.A. Bigdeli, G. Gholami, E. Sheikhhosseini, P-Dodecylbenzenesulfonic acid (DBSA), a |
| [33] | A. Corma, H. Garcia, Organic reactions catalyzed over solid acids, Catal. Today 38 (1997) 257-308. |
| [34] | B. Karimi, D. Zareyee, A high loading sulfonic acid-functionalized ordered nanoporous silica as an efficient and recyclable catalyst for chemoselective deprotection of tert-butyldimethylsilyl ethers, Tetrahedron Lett. 46 (2005) 4661-4665. |
| [35] | B. Karimi, M. Khalkhali, Solid silica-based sulfonic acid as an efficient and recoverable interphase catalyst for selective tetrahydropyranylation of alcohols and phenols, J. Mol. Catal. A: Chem. 232 (2005) 113-117. |
| [36] | I.K. Mbaraka, D.R. Radu, V.S. Lin, B.H. Shanks, Organosulfonic acid-functionalized mesoporous silicas for the esterification of fatty acid, J. Catal. 219 (2003) 329-336. |
| [37] | K. Wilson, A.F. Lee, D.J. Macquarrie, J.H. Clark, Structure and reactivity of sol-gel sulphonic acid silicas, Appl. Catal. A: Gen. 228 (2002) 127-133. |
| [38] | X.L. Shi, H.X. Yang, M.L. Tao, W.Q. Zhang, Sulfonic acid-functionalized polypropylene fiber: highly efficient and recyclable heterogeneous |
| [39] | J. Peng, Y. Deng, Ionic liquids catalyzed Biginelli reaction under solvent-free conditions, Tetrahedron Lett. 42 (2001) 5917-5919. |
| [40] | R. Ghosh, S. Maiti, A. Chakraborty, In(OTf)3-catalysed one-pot synthesis of 3,4-dihydropyrimidin-2(lH)-ones, J. Mol. Catal. A: Chem. 217 (2004) 47-50. |