



 , Dong-Mei Wangb, Yuan-Feng Tongb, Song Wub, Hai-Wei Huangc, Bao-Ming Ningc
, Dong-Mei Wangb, Yuan-Feng Tongb, Song Wub, Hai-Wei Huangc, Bao-Ming Ningc
b Institute of Materia Medica, Chinese Academy of Medical Sciences & Peking Union Medical College, Beijing 100050, China;
c National Institutes for Food and Drug Control, Beijing 100050, China
Methotrexate ((2S)-2-[[4-[[(2,4-diaminopteridin-6-yl)methyl]- methylamino]benzoyl]amino]pentanedioic acid; MTX) is an antineoplastic therapeutic medicine that acts as an anti metabolite of folic acid [1, 2, 3]. It is mainly used in the treatment of meningeal leukemia,psoriasis,and rheumatoid arthritis. The most common adverse effects of MTX include ulcerative stomatitis,low white blood cell count,nausea,abdominal pain,fatigue,fever,dizziness, acute pneumonitis,and rarely,pulmonary fibrosis [3, 4].
At present,the impurity limit for MTX has definite provisions in the pharmacopoeia of each country [1, 5, 6, 7]. For example,in the United States Pharmacopeia and Chinese Pharmacopeia,the total amounts of all impurities should not exceed 2.0% while each single impurity should not exceed 0.5%. In the European Pharmacopoeia (EP) and British Pharmacopoeia (BP),twelve impurities are specified (impurities A–L). The EP and BP both set specific limits for each impurity. In detail,impurityCis less than0.5%,impurities Band E are less than 0.3%,impurities H and I are less than 0.2%,and the other impurities (impurities A,D,F,G,J,K,L) are less than 0.05%. In addition,the sum of impurities,other than B,C,and E,is less than 0.5%. In our previous study of impurities in MTX [8],we found that the impurity profile of MTX drug substances sold in China differed from the reported profiles. We also detected a new impurity, methotrexate 5-ethyl ester,which is not included in the EP or BP. Surprisingly,this impurity was the most abundant impurity in some MTX drug substances. Herein,it is necessary to develop a method to determine impurities in MTX drug substances for improving the quality control standards.
To date,several methods have been described for testing MTX in bulk material,in pharmaceutical dosage forms and in biological samples. These methods included,high-performance liquid chromatography (HPLC) [1, 5, 6, 7, 9, 10, 11, 12],capillary electrophoresis [13],high-performance liquid chromatography tandem mass spectrometry (HPLC–MS) methods [8, 14, 15]. However,very few methods have been described in the context of testing impurities in MTX [1, 2, 5, 6, 7, 8, 9],and the run-time of existing method is commonly very long. Due to ultra-performance liquid chromatography (UPLC) with high separation resolution and rapid analysis characterization,a simple and rapid UPLC method was developed to simultaneously determine the sixmain impurities of MTX drug substances,namely N-methylfolic acid (impurity C in EP and BP),4-amino-N10-methylpteroic acid (impurity E in EP and BP),N10-methylpteroic acid (impurity D in EP and BP),methotrexate 5-methylester (impurity H in EP and BP),methotrexate dimethyl ester (impurity J in EP and BP) andmethotrexate 5-ethyl ester (termed impurity N,newimpurity). To our knowledge,this is the first report describing the use of UPLC to detect impurities in MTX drug substances. The method was validated in accordance with the International Conference on Harmonisation of Technical Registration of Pharmaceuticals for Human Use (ICH) Guidelines [16],and was successfully used to detect MTX impurities in drug substances. 2. Experimental
2.1. Instruments and materials
The assay was performed on a LC-20A system (Shimadzu, Kyoto,Japan),comprising a LC-20AD XR pump,a SIL-20A XR autosampler,a CTO-20A column oven,and a SPD-M20A UV detector. LC solution software was used for LC control and data acquisition (Kyoto,Japan).
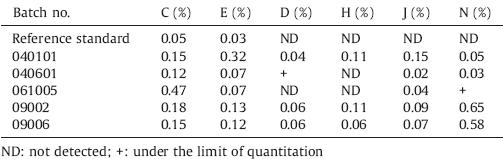
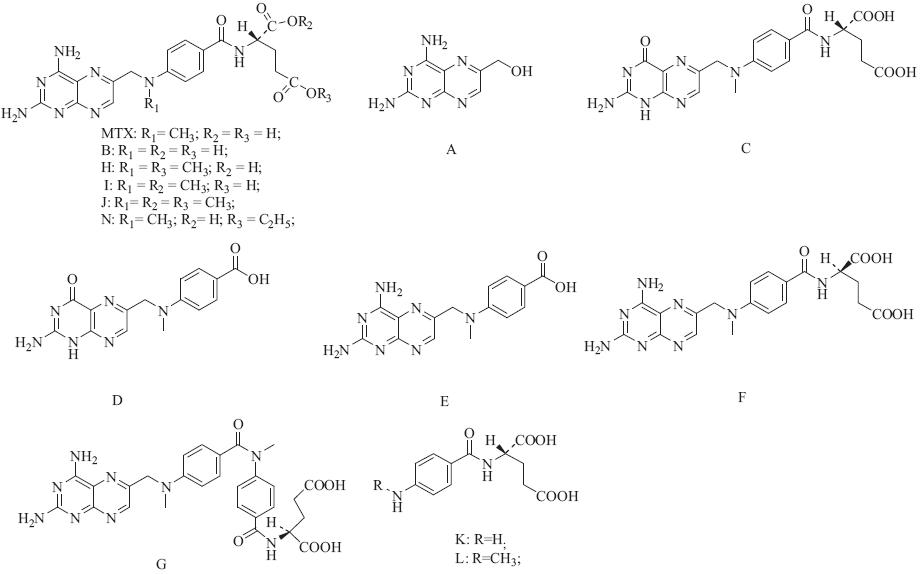
The structures of the six impurities are shown in Fig. 1. Five batches of MTX drug substances (batches 09002,09006,040601, 040101,and 061005) were obtained from four different companies. Batches 09002 and 09006 were obtained from market sold in China. The other three batches were provided by the National Institutes for Food and Drug Control and were collected from two different companies (batch 040601 and 040101 were from same company). The MTX reference standard was obtained from the National Institutes for Food and Drug Control (purity: 99.4%). The MTX impurities reference standards were synthesized by Professor Song Wu using organic chemistry methods and their structures were confirmed based on their MS,1H NMR and 13C NMR data. The MTX impurities reference standards were separated and purified in our lab. The purity of these compounds was more than 95% (determined by HPLC).

|
Download:
|
| Fig. 1.Chemical structures of 13 impurities and MTX. The letters assigned to A–L are identical to EP and BP,while impurity N is a new impurity. | |
Acetonitrile of LC/MS reagent grade was obtained from Mallinckrodt Baker,Inc. (Phillipsburg,NJ,USA). Deionized water was purified using a Millipore water purification system (Millipore, Billerica,MA,USA). Analytical grade acetic acid was obtained from Merck Inc. (Darmstadt,Germany). Sodium dihydrogen phosphate and phosphoric acid of analytical grade were purchased from Beijing Chemical Corp. (Beijing,China). Analytical grade dimethylsulfoxide (DMSO) was obtained from Acros Organics Corp. (Geel,Belgium) 2.2. UPLC analytical conditions
Chromatographic separation was performed using an Agilent Zorbax Extend-C18 column (4.6 mm × 50 mm,1.8 mm). The mobile phase consisted of solvent A (deionized water with 20 mmol/L sodium dihydrogen phosphate,adjusted to pH 3.0 with phosphoric acid) and solvent B (acetonitrile),delivered at a flow rate of 2.2 mL/min. Gradient elution was started at 8% B, which was held until 4.0 min,followed by an increase to 12% B at 9 min,and maintained until 11 min. The post-run time is 5 min. The column temperature was maintained at 40°C and the sample injection volume was 3 mL. Detection was carried out at 305 nm. 2.3. Preparation of system suitability solution
Standard stock solutions of the six impurities were prepared in DMSO at a concentration of 500 mg/mL,respectively. Appropriate amounts of the stock solutions were then mixed and diluted with DMSO to prepare working solutions with a desired concentration about 25 mg/mL. Additionally,25 mg of the MTX reference standard were accurately weighed,transferred to a 5 mL volumetric flask,dissolved and diluted to the volume with the working solution (25 mg/mL) of six impurities. 2.4. Sample preparation
A 25 mg sample of MTX drug substances were accurately weighed and transferred to a 5 mL volumetric flask then dissolved and diluted to the volume with DMSO to obtain the test solution with the concentration at 5 mg/mL. Each test solution was filtered through a 0.45 mm nylon filter film before analysis. 3. Results and discussion
For sample preparation,we first dissolved the drug substance samples in ammonia diluted 100 times with deionized water. However,we subsequently found that the esterified impurities of MTX(i.e.,impurity H,J,N)werehydrolyzedindiluteammonia,based on degradation studies under basic and acidic conditions which decreased their concentrations. These studies are described in our previous impurity profile study [8]. For these reasons,we used DMSO as the solvent for dissolving the MTX drug substances and reference standards. In addition,in order to obtain chromatograms with good resolution of the impurities andMTX,theUPLCconditions were optimized using the impurity standards mixture and the MTX reference standardsolutions.We testeddifferent columns,including the Agilent Zorbax Extend-C18 (4.6mm× 50mm,1.8 mm),Agilent Zorbax SB-C18 (4.6mm× 50mm,1.8 mm),Agilent Zorbax Eclipse XDB-C18 (4.6mm× 50mm,1.8 mm),Phenomenex Kinetex C18 (4.6mm× 100 mm,2.6 mm),and finally selected the Agilent Zorbax Extend-C18 (4.6mm× 50mm,1.8 mm). After different mobile phases consist of acetonitrile or methanol as organic solvent and deionized water containing sodium dihydrogen phosphate,ammoniumformate, formic acidwere tried,itwas foundthatmobile phase consisting of solvent A (deionized water with 20mmol/L sodium dihydrogen phosphate,adjusted topH3.0withphosphoric acid) and solvent B (acetonitrile) could improve the separation resolution. A gradient elution program was necessary to rapidly separate six impurities andMTX. Good and rapid separation of six impurities and MTX was achieved under the specified UPLC conditions within 16 min. Representative UPLC chromatograms are shown in Fig. 2.

|
Download:
|
| Fig. 2.Representative chromatograms for using UPLC method system suitability solution (A) and MTX chemical reference standard (B). The numbers assigned to compounds are the same as those in Fig. 1. | |
The validation approach was executed under the ICH guidelines for validation of analytical procedures [16]. Key analytical parameters, including specificity,LOD,LOQ,linearity,precision,and recovery were evaluated. All calibration curves displayed good linearity (r > 0.999)within the testedconcentrationranges.The LOD and LOQ of the six analytes were all less than 0.774 mg/mL and 1.03 mg/mL. The relative standard deviation (RSD) for intra- and inter-day precision of the six analytes was less than 9.8%,including at the LOQ. Themean average recoveries ranged from95.2% to 103%, except at the LOQ,where it ranged from 82.7% to 117% (Table 1).
| Table 1 Method validation results. |

The validated UPLC method was applied to the analysis of five batches of MTX drug substances from four different companies. Unlike previous reported assays,our method was the first report to detect the ethyl esterified impurity in drug substances. The results in Table 2 indicate that the levels ofMTX impurities differed among batches,probably because of differences in the synthetic processes and environmental conditions. According to our results,only two batches of drug substances met the limits standard of impurity in EP or BP [1, 5]. However,in batches 09002,09006,and 040101,the levels of two esterified impurities,J and N,were higher than the maximum permitted level. In addition,the level of impurity D was also above the maximum permitted level in two batches (batches 09002 and 09006). Therefore,the impurity content should be determined in multisource drug substances to confirm the quality and safety of marketed drugs.
| Table 2 Assay of six impurities in MTX reference standard and five MTX drug substances. |
{kind=link}
{kind=link}
In this paper,an UPLC method for the simultaneous determination of impurities in MTX drug substances was developed and validated. The established method was simple,rapid,sensitive,and reliable. We believe that this convenient method can be applied for the routine monitoring and quality control of impurities in MTX drug substances. To our knowledge,this was the first report to detect the contents of methotrexate 5-ethyl ester (termed impurity N) in MTX drug substances. Acknowledgments
We thank the Ministry of Public Health of the People’s Republic of China (No. 200802038) and the Ministry of Science and Technology of the People’s Republic of China (No. 2011IM030200) for financial support of this work.
| [1] | European Pharmacopoeia, European Directorate for the Quality of Medicines & Health Care, 7th ed., European Pharmacopoeia, Strasbourg, 2010, pp. 2467-2469. |
| [2] | R. Gotti, D.A. El-Hady, V. Andrisano, et al., Determination of the chiral and achiral related substances of methotrexate by cyclodextrin-modified micellar electrokinetic chromatography, Electrophoresis 25 (2004) 2830-2837. |
| [3] | Methotrexate, Wikipedia, http://en.wikipedia.org/wiki/Methotrexate. |
| [4] | S. Shen, T. O'Brien, L.M. Yap, H.M. Prince, C.J. McCormack, The use of methotrexate in dermatology: a review, Australas. J. Dermatol. 53 (2012) 1-18. |
| [5] | The Stationery Office, The British Pharmacopoeia Commission British Pharmacopoeia, The Stationery Office, 2012, pp. 1427-1429. |
| [6] | The United States Pharmacopeial Convention, The 35th revision of the United States Pharmacopeia (USP 35) and the 30th edition of the National Formulary (NF 30), Rockville, MD, 2011, pp. 3855-3857. |
| [7] | The Pharmacopoeia Commission of PRC, The Pharmacopoeia of the People's Republic of China. Part Ⅱ, China Medical Science and Technology Press, Beijing, 2010p. 149. |
| [8] | C.S. Wu, Y.F. Tong, P.Y. Wang, et al., Identification of impurities in methotrexate drug substances using high-performance liquid chromatography coupled with a photodiode array detector and Fourier transform ion cyclotron resonance mass spectrometry, Rapid Commun. Mass Spectrom. 27 (2013) 971-978. |
| [9] | D.A. El-Hady, N.A. El-Maali, R. Gotti, et al., Methotrexate determination in pharmaceuticals by enantioselective HPLC, J. Pharm. Biomed. Anal. 37 (2005) 919-925. |
| [10] | K. Michail, M.S. Moneeb, Determination of methotrexate and indomethacin in urine using SPE-LC-DAD after derivatization, J. Pharm. Biomed. Anal. 55 (2011) 317-324. |
| [11] | M. Uchiyama, T. Matsumoto, T. Matsumoto, et al., Simple and sensitive HPLC method for the fluorometric determination of methotrexate and its major metabolites in human plasma by post-column photochemical reaction, Biomed. Chromatogr. 26 (2012) 76-80. |
| [12] | S. Fang, C.P. Lollo, C. Derunes, M.J. LaBarre, Development and validation of a liquid chromatography method for simultaneous determination of three process-related impurities: yeastolates, triton X-100 and methotrexate, J. Chromatogr. B Analyt. Technol. Biomed. Life Sci. 879 (2012) 3612-3619. |
| [13] | H.L. Cheng, S.S. Chiou, Y.M. Liao, et al., Analysis of methotrexate and its eight metabolites in cerebrospinal fluid by solid-phase extraction and triple-stacking capillary electrophoresis, Anal. Bioanal. Chem. 398 (2010) 2183-2190. |
| [14] | G. Chen, J.P. Fawcett, M. Mikov, et al., Simultaneous determination of methotrexate and its polyglutamate metabolites in Caco-2 cells by liquid chromatographyandem mass spectrometry, J. Pharm. Biomed. Anal. 50 (2009) 262-266. |
| [15] | E. den Boer, S.G. Heil, B.D. van Zelst, et al., A U-HPLC-ESI-MS/MS-based stable isotope dilution method for the detection and quantitation of methotrexate in plasma, Ther. Drug Monit. 34 (2012) 432-439. |
| [16] | ICH Guidelines, Impurities in new drug substances Q3A(R2), October 2006. |