




In the past several decades,a series of 20,30-dideoxynucleosides (ddNs),including zidovudine (AZT),didanosine (ddI),zalcitabine (ddC),stavudine (d4T),lamivudine (3TC),and abacavir (ABC),have been licensed for the treatment of acquired immunodeficiency syndrome (AIDS) targeting human immunodeficiency virus reverse transcriptase (HIV-RT) [1]. In the host cells,the transformation of ddNs into the 50-triphosphate form is essential for their integration into viral DNA as chain terminators [2]. Therefore,ddN 5'-triphosphates have always been important synthetic targets for the investigation of the antiviral mechanisms of the parent ddNs [3]. AZT 5'-triphosphate (AZTTP) and d4T 5'-triphosphate (d4TTP) have been prepared via the conventional ‘‘one-pot,three-step’’ [4] and salicyl chlorophosphite (SCP) methods in low to moderate yields [5]. Establishment of facile and efficient synthetic methods for ddN 5'-triphosphates remains a huge challenge for phosphorus chemists.
It has been reported that ddN-induced decrease in nucleoside kinase activity is a major cause for the reduced therapeutic efficacy and drug resistance in HIV therapy [6]. To overcome these clinical limitations,homodimers of ddN 5'-monophosphates have been synthesized as prodrugs in different laboratories. For instance, Magnani et al. successfully encapsulated the homodinucleotide of AZT (AZTp2AZT) into the erythrocytes to target macrophages [7]. Herdewijn et al. demonstrated that did4T pyrophosphate (d4Tp2d4T) could serve as an effective substrate of HIV-RT [8]. These homodimers of nucleoside 50-monophosphate (NMP) have been synthesized by reacting one molecule of NMP with another molecule of activated NMP such as P1-AZT-5'-P2-diphenyl pyrophosphate [6] and d4T 5'-phosphorimidazolide in low yields [8]. Recently,Parang et al. reported a solid-phase approach for the synthesis of AZTTP and AZTp2AZT [9]. However,the complicated procedures for the preparation of polymer-bound reagents limit its applications. Herein,we report a one-pot protocol for the synthesis of both triphosphates and homodinucleotides of ddNs from the corresponding nucleoside 5'-H-phosphonates. 2. Experimental
All reactions were performed in anhydrous solvents under an atmosphere of dry argon. Ion exchange chromatography employed DEAE Sephadex A-25 exchanger. NMR spectra were obtained on a 400 MHz instrument with chemical shifts reported in parts per million (ppm,δ). IR spectra were recorded on an FT-IR spectrometer. Low-resolution mass spectra were obtained with an ion trap mass spectrometer and reported as m/z. 2.1. General procedure for the synthesis of nucleoside 5'-Hphosphonate monoesters (2a-b)
To a solution of PCl3 (3.2 mL,35 mmol) in CH2Cl2 (100 mL) at -20°C was added nucleoside (3.5 mmol). The reaction was kept at -20°C for 1 h and stirred for 4 h at room temperature. The solution was concentrated in vacuo,and the residue was treated with 10% aqueous Et3 (4 mL) for 5 min. The crude product was obtained after the solvent was removed under reduced pressure. Flash column chromatography on silica gel (CH2Cl2/MeOH 20:1 to 5:1 with 0.5% Et3) afforded the nucleoside 5'-H-phosphonate as a triethylammonium salt,a glassy solid.
AZT-5'-H-phosphonate monoester,triethylammonium salt (2a): 1.30 g,86%; 1H NMR (400 MHz,D2O): d 7.66 (s,1H,H6), 6.75 (d,1H,JP,H = 636.4 Hz),6.19 (t,1H,J = 6.8 Hz,H1'),4.46 (m,1H, H3'),4.13 (m,1H,H4'),4.06 (m,2H,H5'),3.16 (q,6H,N(CH2CH3)3), 2.45 (t,1H,J = 6.0 Hz,H2O),1.87 (s,3H,CH3),1.24 (t,9H, N(CH2CH3)3); 13C NMR (100 MHz,D2O): d 166.4,151.6,137.3, 111.6,85.1,83.1 (d,3JC,P = 8.0 Hz),63.1 (d,2JC,P = 4.1 Hz),60.6,46.8, 36.4,11.7,8.4; 31P NMR (162 MHz,D2O): δ 6.35 (d,1P, JP,H = 637 Hz); IR (KBr,cm-1): νmax 3727,3425,2938,2805, 2742,2676,2491,2394,2351,2108,1708,1474,1397,1273,1086, 1057,966,551,420; LRMS (ESI-): calcd. for C10H13N5O6p (m/z): 330.1 [M–H]-; found: 330.0.
d4T-5'-H-phosphonate monoester,triethylammonium salt (2b): 1.21 g,89%; 1H NMR (400 MHz,D2O): δ 7.57 (s,1H,H6), 6.92 (s,1H,H1'),6.67 (d,1H,JP,H = 637.6 Hz),6.46 (d,1H,J = 5.6 Hz, H2O),5.95 (d,1H,J = 6.4 Hz,H3'),5.06 (m,1H,H4'),4.04 (m,2H, H5'),3.18 (q,6H,N(CH2CH3)3),1.86 (s,3H,CH3),1.26 (t,9H, N(CH2CH3)3); 13C NMR (100 MHz,D2O): δ 166.8,152.4,138.4, 134.4,125.6,111.4,90.2,86.0 (d,3JC,P = 7.4 Hz),64.1 (d, 2JC,P = 4.1 Hz),46.8,11.6,8.4; 31P NMR (162 MHz,D2O): δ 6.37 (d,1P,JP,H = 638 Hz); IR (KBr,cm-1): νmax 3731,3415,3130,2939, 2810,2742,2677,2491,2388,2360,1692,1473,1397,1218,1080, 988,910,836,779,577; LRMS (ESI-): calcd. for C10H12N2O6P (m/z): 287.0 [M–H]-; found: 287.1. 2.2. General procedure for the synthesis of homonucleotides (3a-b)
To a flask containing H-phosphonate (0.1 mmol) pyridine (1.0 mL) and TMSCl (40 mL,0.3 mmol) were added. After 5 min, a solution of I2 in pyridine (0.2 mol/L,600 mL) was added dropwise via a microsyringe. Addition of the I2 solution continued until the color of iodine stopped fading. The brown solution was stirred for 5 min at room temperature. H2O in DMF (36 mL,10% (v/v)) was quickly injected to the reaction solution. After 5 min,the second portion of H2O in DMF (18 mL,10% (v/v)) was added. The reaction was stirred for 10 min and concentrated in vacuo. The crude product was dissolved in deionized H2O (0.5 mL) and loaded onto a DEAE Sephadex A-25 ion exchange column (1.6 × 25 cm). Elution with TEAB buffer (0.2–0.5 mol/L),combination of the appropriate fractions,and lyophilization afforded the product as a triethylammonium salt. Passage of the solution of triethylammonium salt in deionized H2O through a bed of Dowex 50W-X8 ion exchange resin (Na+ form) and lyophilization afforded the homodinucleotide as a disodium salt,a white solid.
P1,P2-diAZT-5'-diphosphate,disodium salt (3a): 25 mg,70%; 1H NMR (400 MHz,D2O): δ 7.66 (s,2H,H6),6.18 (t,2H,J = 6.4 Hz, H1'),4.47 (m,2H,H3'),4.25–4.05 (m,6H,H4',H5'),2.44 (t,4H, J = 6.4 Hz,H2O),1.88 (s,6H,CH3); 13C NMR (100 MHz,D2O): δ 166.6, 151.7,137.5,111.8,84.8,82.9,65.6,60.6,36.4,11.8; 31P NMR (162 MHz,D2O): δ –11.26 (s); IR (KBr,cm-1): νmax 3723,3530, 2941,2819,2379,2350,2316,2113,1708,1480,1368,1276,1124, 1062,957,518,421; LRMS (ESI–): calcd. for C20H25N10O13P2 (m/z): 675.1 [M–H]-; found: 675.2. P1,P2-did4T-5'-diphosphate,disodium salt (3b): 24 mg,75%; 1H NMR (400 MHz,D2O): δ 7.56 (s,2H,H6),6.86 (s,2H,H1'),6.40 (d,2H,J = 4.4 Hz,H2O),5.86 (d,2H,J = 4.8 Hz,H3'),4.99 (m,2H,H4'), 4.06 (m,4H,H5'),1.81 (s,6H,CH3); 13C NMR (100 MHz,D2O): δ 166.7,152.2,138.3,134.2,125.6,111.5,90.0,85.9,66.5,11.5; 31P NMR (162 MHz,D2O): δ –11.50 (s); IR (KBr,cm-1): νmax 3729, 3566,3414,3128,2944,2886,2815,2379,2351,2315,1706,1478, 1400,1365,1249,1122,1048,956,913,839,782,590; LRMS (ESI-): calcd. for C2OH23N4O13P2 (m/z): 589.1 [M–H]-; found: 589.2. 2.3. General procedure for the synthesis of nucleoside 5'- triphosphates (4a–b)
Prior to the reaction,tris(tetra-n-butylammonium) hydrogen pyrophosphate (0.3 mmol) and H-phosphonate (0.1 mmol) were dried under vacuum in two separate flasks. The H-phosphonate was oxidized according to the procedure described in the synthesis of 3a-b. Pyrophosphate in DMF (0.5 mL) was quickly injected into the reaction solution. The reaction was stirred for 20 min and concentrated in vacuo. The residue was dissolved in cold deionized water (1 mL) with sonication. The precipitated iodine was removed by filtration. Cold deionized H2O (1 mL) was added to the flask and filtered through a microfilter. The combined filtrate was concentrated in vacuo. The residue was dissolved in a NaOAc aqueous solution (10 mol/L,0.4 mL). After EtOH (20 mL) was added,the white precipitate was collected by centrifuge. The crude product was dissolved in deionized H2O (0.5 mL) and loaded onto a DEAE Sephadex A-25 ion exchange column (1.6 cm × 25 cm). Elution with TEAB buffer (0.2–0.5 mol/L),combination of the appropriate fractions,and lyophilization afforded the product as a triethylammonium salt. Passage of the solution of the triethylammonium salt in deionized H2O through a bed of Dowex 50W-X8 ion exchange resin (Na+ form) and lyophilization afforded the nucleoside 5'-triphosphate as a tetrasodium salt,a white solid.
AZT-5'-triphosphate,tetrasodium salt (4a): 29 mg,48%; 1H NMR (400 MHz,D2O): δ 7.73 (s,1H,H6),6.26 (t,1H,J = 6.8 Hz,H1'),4.57 (m,1H,H3'),4.21 (m,3H,H4',50),2.47 (m,2H, H2O),1.91 (s,3H,CH3); 13C NMR (100 MHz,D2O): δ 166.5,151.8, 137.2,111.8,84.9,83.0 (d,3JC,P = 9.2 Hz),65.7 (d,2JC,P = 5.5 Hz), 60.9,36.2,11.6; 31P NMR (162 MHz,D2O): δ –9.08 (d,JP,P = 19.9 Hz, 1P),–11.65 (d,JP,P = 20.0 Hz,1P),–23.17 (t,JP,P = 20.0 Hz,1P); IR (KBr,cm-1): νmax 3731,3596,2936,2810,2376,2347,2315,2112, 1708,1481,1368,1278,1128,1064,976,517,419; LRMS (ESI-): calcd. for C>10H15N5O13P3 (m/z): 506.0 [M–H]-; found: 506.1. d4T-5'-triphosphate,tetrasodium salt (4b): 29 mg,53%; 1H NMR (400 MHz,D2O): δ 7.59 (s,1H,H6),6.94 (d,1H,J = 1.2 Hz,H1'),6.52 (d,1H,J = 6.4 Hz,H2O),5.93 (d,1H,J = 6.0 Hz,H3'), 5.10 (m,1H,H4'),4.17 (m,2H,H5'),1.87 (s,3H,CH3); 13C NMR (100 MHz,D2O): δ 166.8,152.3,138.1,134.3,125.1,111.5,90.0, 85.9 (d,3JC,P = 8.2 Hz),66.5 (d,2JC,P = 5.1 Hz),11.4; 31P NMR (162 MHz,D2O): δ –7.90 (d,JP,P = 17.3 Hz,1P),–11.18 (d, JP,P = 19.4 Hz,1P),–22.21 (t,1P); IR (KBr,cm-1): νmax 3730, 3490,3126,2940,2819,2378,2349,2317,1701,1478,1401,1265, 1110,1047,989,931,837,783,596; LRMS (ESI–): calcd. for C10H14N2O13P3 (m/z): 463.0 [M–H]-; found: 463.1. 3. Results and discussion
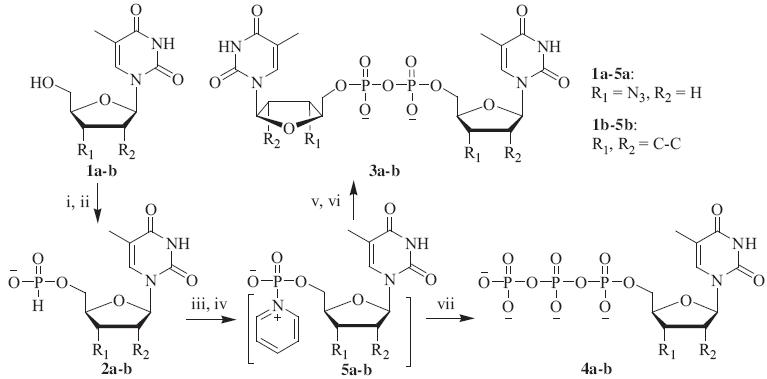
As shown in Scheme 1,the H-phosphonates of AZT and d4T (2a-b) were efficiently prepared by treating the antiviral ddNs (1a–b) with 10.0 equiv. of PCl3 in CH2Cl2. After excess PCl3 was removed by evaporation,the dichlorophosphites were hydrolyzed to yield the corresponding H-phosphonates. Comparing with other phosphitylating reagents,such as tri(imidazolyl)phosphite (PIm3) [10],diphenyl phosphite (DPP) [11],and salicyl chlorophosphite (SCP) [12],PCl3 afforded 2a-b in higher yields ranging from86-89%.

|
Download:
|
| Scheme 1.Synthesis of AZT and d4T homodinucleotides (3a–b) and triphosphates (4a–b). Reagents and conditions: (i) PCl3 (10.0 equiv.),CH2Cl2,-20°C,1 h,RT,4 h; (ii) H2O, Et3N,10 min (86% for 2a/89% for 2b); (iii) TMSCl (3.0 equiv.),pyridine,5 min; (iv) I2 (1.2 equiv.),5 min; (v)H2O (1.0 equiv.),5 min; (vi) H2O (0.5 equiv.),10 min (70% for 3a/75% for 3b); (vii) (nBu4N)3HP2O7 (3.0 equiv.),20 min (48% for 4a/53% for 4b). | |
{kind=link}
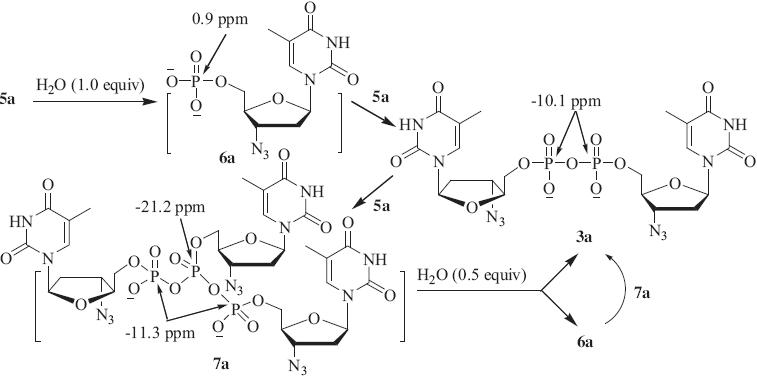
Based on reported procedures [11, 12, 13],2a-b were converted into the highly reactive pyridinium phosphoramidate intermediates by treating the H-phosphonates with 3.0 equiv. of TMSCl, followed by the addition of 1.2 equiv. of I2. 31P NMR tracing experiment showed that upon the addition of 1.0 equiv. of H2O in DMF (10%,v/v),the zwitterionic phosphoramidate intermediate (5a) was transformed into the P1,P2-diAZT-5'-diphosphate (3a,-10.1 ppm) and P1,P2,P3-triAZT-5'-triphosphate (7a,-11.3 and-21.2 ppm with a 2:1 ratio) in 5 min. When the second portion of 0.5 equiv. of H2O was added,7a was partially hydrolyzed to give AZT 50-monophosphate (6a,0.9 ppm) and 3a. A large portion of 6a in turn quickly reacted with 7a to give two molecules of desired 3a (Scheme 2). The result of the 31P NMR tracing experiment (Fig. 1) confirmed the existence of P1,P2,P3-trinucleoside triphosphate intermediate [14] and reaction path proposed in our previous report [11]. According to the established sequential hydrolysis protocol,the homodinucleotides of AZT and d4T (3a-b) were obtained in 70–75% isolated yields (Scheme 1).

|
Download:
|
| Scheme 2.Proposed reaction path illustrating measured 31P chemical shifts of intermediates and products for the sequential conversion of phosphoramidate intermediate (5a) to AZTp2AZT (3a). | |
{kind=link}

|
Download:
|
| Fig. 1.Stacked 31P NMR spectra of the reaction mixture prior to and after the sequential addition of H2O. (a) Phosphoramidate intermediate (5a); (b) 5 min after the first addition of H2O (1.0 equiv.); (c) 5 min after the second addition of H2O (0.5 equiv.); (d) 10 min after the second addition of H2O (0.5 equiv.). | |
{kind=link}
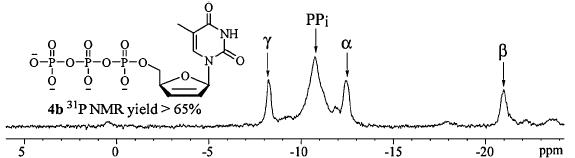
Alternatively,intermediate 5a could be treated with pyrophosphate to generate the triphosphates of AZT and d4T (4a–b) [12]. However,addition of 1.0 equiv. of pyrophosphate according to the reported procedure only afforded 4a–b in low yields (<20%), because the zwitterionic phosphoramidates of ddNs were more prone to form dinucleoside polyphosphate byproducts compared to their ribonucleoside counterparts. To solve this problem,a larger excess of pyrophosphate reagent was added to improve the coupling specificity [15]. 31P NMR tracing experiment revealed that 3.0 equiv. of pyrophosphate could effectively increase the yield of 4b to 65% (Fig. 2). Ion exchange chromatography purification afforded triphosphates 4a–b in 48 and 53% yields, respectively (Scheme 1).

|
Download:
|
| Fig. 2.The 31P NMR spectrum of the crude reaction mixture of d4TTP (4b). | |
{kind=link}
In summary,we developed a one-pot method for the synthesis of the triphosphates and homonucleotides of antiviral ddNs from the corresponding nucleoside 5'-H-phosphonate precursors. This method features easily accessible starting materials,short reaction time,and good isolated yields and can be applied to a variety of antiviral nucleoside analogs. Acknowledgments
Wethank the NationalNatural Science Foundation of China (Nos. 21002041 and 21262014),Natural Science Foundation of Jiangxi Province (No. 20114BAB203008),Project of the Science Funds of Jiangxi Education Office (No. GJJ12589),Key Project of Chinese Ministry of Education (No. 212092),and Scientific Research Foundation of Chinese Ministry of Human Resources and Social Security for Returned Chinese Scholars for financial support.
| [1] | (a) E. de Clercq, P. Herdewijn, Strategies in the design of antiviral drugs, Nat. Rev. Drug Discov. 1 (2002) 13-25; (b) E. de Clercq, Molecular targets for antiviral agents, J. Pharmacol. Exp. Ther. 297 (2001) 1-10. |
| [2] | B.Öberg, Rational design of polymerase inhibitors as antiviral drugs, Antivir. Res. 71 (2006) 90-95. |
| [3] | (a) E. de Clercq, J. Neyts, Antiviral agents acting as DNA or RNA chain terminators, Handb. Exp. Pharmacol. 189 (2009) 53-84; (b) X.J. Liu, W. Xie, R.H. Huang, Structure-based design, synthesis, and in vitro assay of novel nucleoside analog inhibitors against HIV-1 reverse transcriptase, Bioorg. Med. Chem. Lett. 15 (2005) 3775-3777. |
| [4] | (a) Q. Ma, I.C. Bathurst, P.J. Barr, G.L. Kenyon, New thymidine triphosphate analogue inhibitors of human immunodeficiency virus-1 reverse transcriptase, J. Med. Chem. 35 (1992) 1938-1941; (b) A. Fatima, C. Christophe, G. Jacques, G. Roger, D. Daniele, The production and evaluation of antibodies for enzyme immunoassay of AZTTP, Nucleos. Nucleot. Nucl. 20 (2001) 243-250; (c) M.M. Mansuri, J.E. Starrett, I. Ghazzouli, et al., 1-(2,3-Dideoxy-b-D-glyceropent-2-enofuranosyl)thymine. A highly potent and selective anti-HIV agent, J. Med. Chem. 32 (1989) 461-466. |
| [5] | J. Ludwig, F. Eckstein, Rapid and efficient synthesis of nucleoside 5'-O-(1-thiotriphosphates), 5'-triphosphates, and 2',3'-cyclophosphorothioates using 2-chloro-4H-1,3,2-benzodioxaphosphorin-4-one, J. Org. Chem. 54 (1989) 631-635. |
| [6] | M. Mauro, C. Anna, F. Alessandra, et al., Synthesis and targeted delivery of an azidothymidine homodinucleotide conferring protection to macrophages against retroviral infection, Proc. Natl. Acad. Sci. USA 93 (1996) 4403-4408. |
| [7] | L. Rossi, S. Serafini, P. Franchetti, et al., Targeting nucleotide dimers containing antiviral nucleosides, Curr. Med. Chem. Anti-Infect. Agents 4 (2005) 37-54. |
| [8] | S.Q. Yang, C. Pannecouque, P. Herdewijn, Synthesis and in vitro enzymatic and antiviral evaluation of d4T polyphosphate derivatives as chain terminators, Chem. Biodiv. 9 (2012) 2186-2194. |
| [9] | Y. Ahmadibeni, K. Parang, Selective diphosphorylation dithiodiphosphorylation triphosphorylation and trithiotriphosphorylation of unprotected carbohydrates and nuclesides, Org. Lett. 7 (2005) 5589-5592. |
| [10] | I. Lindh, J. Stawinski, A general method for the synthesis of glycerophospholipids and their analogs via H-phosphonate intermediates, J. Org. Chem. 54 (1989) 1338-1342. |
| [11] | Q. Sun, S. Liu, J. Sun, et al., One-pot synthesis of symmetrical P1,P2-dinucleoside-50-diphosphates from nucleoside-5'-H-phosphonates: mechanistic insights into reaction path, Tetrahedron Lett. 54 (2013) 3842-3845. |
| [12] | Q. Sun, J.P. Edathil, R. Wu, et al., One-pot synthesis of nucleoside 5'-triphosphates from nucleoside 50-H-phosphonates, Org. Lett. 10 (2008) 1703-1706. |
| [13] | R. Wu, E.D. Smidansky, H.S. Oh, et al., Synthesis of a 6-methyl-7-deaza analogue of adenosine that potently inhibits replication of polio and dengue viruses, J. Med. Chem. 53 (2010) 7958-7966. |
| [14] | (a) D.G. Knorre, A.V. Lebedev, A.S. Levina, A.I. Rezvukhin, V.F. Zarytova, Active monomeric nucleotide intermediate in the oligonucleotide synthesis, Tetrahedron 30 (1974) 3073-3079; (b) M. Sekine, M. Ushioda, T. Wada, K. Seio, Synthesis of TMG-capped RNA-DNA chimeric oligonucleotides, Tetrahedron Lett. 44 (2003) 1703-1707. |
| [15] | Q. Sun, S. Gong, J. Sun, et al., A P(V)-N activation strategy for synthesis of nucleoside polyphosphates, J. Org. Chem. 78 (2013) 8417-8426. |