




With their outstanding electronic and optical properties, phthalocyanines and their metallic derivatives have become a promising family of materials in recent years having already been used in laser printers, photo-copiers, light-emitting diodes (LEDs), data storage systems and photovoltaic cells. Their potential applications in optical limiting, gas sensors, field-effect transistors (FETs) and photodynamic therapy have attracted even more research interests. In the above research, phthalocyanine thin films have always required fabrication. Consequently, there are several ways to fabricate the thin films [1]: Langmuir-Blodgett technique, solvent evaporation, thermal evaporation, electrochemical deposition, stamping and self-assembly method. Among them, the selfassembly method seems to become even more attractive with the growth of the research field in molecular electronics. This method has already been used in fabricating organic electronic devices [2]. Compared with thin films prepared by other methods, the selfassembled monolayers (SAMs) are simple to fabricate, very thermally and mechanically stable and can be grown on substrates of all sizes. The self-assembling behavior of small molecules bearing sulfur-containing substituents, such as disulfide, S-acetyl and mercapto-groups, has been studied extensively [3]. They have been shown to form self-assembled monolayers on gold, silver and copper substrates. Compared to these small molecules, the selfassembly of phthalocyanines with excellent optical and electrical properties may be of more importance. As is well known, a variety of substituents can be introduced onto the periphery of the phthalocyanine rings and their properties can be finely tuned as such. Many substituted phthalocyanines have been synthesized, but only limited work has been done on the synthesis and the selfassembly of phthalocyanines bearing sulfur-containing substituents [4, 5, 6]. We report here on the synthesis and self-assembly of two phthalocyanines bearing sulfur-containing substituents including disulfide and S-acetyl groups.
The precursors, 4-nitrophthalonitrile and 4-tert-butylphthalonitrile, were purchased from Tokyo Kasei Kogyo Co., Ltd. and used as received without further purification. Phthalocyanines 8, 9 and S-(4-iodophenyl)ethanethioate were prepared according to literature methods. Tetrahydrofuran (THF) was distilled from Nabenzophenone under nitrogen. Other solvents were dried over 4Å molecular sieve and distilled prior to use. The 1HNMR spectra were obtained on a Varian Unity 200 NMR Spectrometer, or Bruker DMX 300 NMR Spectrometer. UV-vis spectra were measured using a Hitachi Model U-3010 Spectrophotometer. MS spectra (MALDI-TOF-MS) were obtained on a Bruker BIFLEX ⅡI Mass Spectrometer.
To an ice-cooled solution of 1,6-hexanediol (59 g, 500 mmol), ptoluene sulfuric acid monohydrate (1.6 g, 8.4 mmol) and dihydropyran (4.2 g, 50 mmol) were added with stirring. The mixture was then allowed to warm to room temperature and stirred for 24 h. The resulting mixture was diluted with ether, washed with saturated aqueous potassium carbonate solution and dried with anhydrous potassium carbonate. After solvent evaporation, the remaining white solid was extracted with petroleum ether (60- 90 ℃). Upon removal of the solvent, 6.4 g (63% yield) of 2 was obtained as colorless oil. Data for 2: 1H NMR (200 MHz, CDCl3): δ 1.41 (m, 4H, CH2), 1.55 (m, 8H, CH2), 1.87 (m, 2H, CH2), 3.31-3.57 (m, 2H, OCH2), 3.64 (t, 2H, J = 7.1 Hz, OCH2), 3.70-3.95 (m, 2H, OCH2), 4.58 (t, 1H, J = 7.3 Hz, OCHO).
A mixture of 4-nitrophthalonitrile (5.0 g, 29 mmol), 1,6- hexanediol monotetrahydropyranyl ether 2 (5.8 g, 29 mmol) and anhydrous potassium carbonate (12 g, 87 mmol) in DMF (150 mL) was stirred overnight at 60 ℃. The solvent was removed under reduced pressure. The residue was dissolved with dichloromethane, filtered and purified by column chromatography (silica gel) using dichloromethane as eluant. The product 3 was obtained as yellow oil (7.5 g, 79% yield). Data for 3: 1H NMR (200 MHz, acetone-d6): δ 1.48 (m, 12H, CH2), 1.80 (m, 2H, CH2), 3.40 (m, 2H, OCH2), 3.75 (m, 2H, OCH2), 4.23 (t, 2H, J = 7.1 Hz, OCH2), 4.54 (m, 1H, OCHO), 7.45 (dd, 1H, phenyl), 7.60 (d, 1H, phenyl), 7.94 (d, 1H, phenyl).
Phthalonitrile 3 (7.5 g, 23 mmol) and p-toluenesulfonic acid (PTSA) (0.43 g, 2.3 mmol) were dissolved in ethanol (150 mL). The resulting solution was stirred at 50 ℃ for 10 h at 50 ℃. After removal of the solvent, the residue was purified by column chromatography (silica gel) using dichloromethane as eluant. Phthalonitrile 4 (3.5 g, 63% yield) was obtained as a light yellow solid. Data for 4: 1H NMR (300 MHz, acetone-d6): δ 1.84 (m, 4H, CH2), 1.50 (m, 4H, CH2), 3.65 (t, 2H, J = 7.1 Hz, OCH2), 4.05 (t, 2H, J = 7.1 Hz, OCH2), 4.58 (br, 1H, OH), 7.18 (dd, 1H, phenyl), 7.25 (d, 1H, phenyl), 7.71 (d, 1H, phenyl).
To n-pentanol (70 mL), 4-tert-butylphthalonitrile (6.1 g, 33 mmol) and phthalonitrile 4 (2.7 g, 11 mmol) were added. The mixture was brought to reflux. Then lithium metal (1.0 g, 0.14 mmol) was added in small portions. The reaction was continued for 6 h. The solvent was removed under reduced pressure. The black residue was washed with methanol and further purified by column chromatography (silica gel) using chloroform as eluant. The first band proved to be tetra-tert-butyl phthalocyanine. Then the second band was collected. Upon removal of the solvents, the desired product was obtained as a shining purple solid (1.7 g, 19% yield). Data for 6: 1H NMR (300 MHz, C6D6): δ 2.34 (br, 1H, NH), -2.79 (br, 1H, NH), 1.71 (m, 2H), 1.86 (m, 31H), 2.16 (m, 2H), 3.84 (m, 2H, OCH2), 4.46 (m, 2H, OCH2), 7.20-9.20 (m, 12H, aryl). MALDI-TOF-MS: m/z calcd. for C50H54N8O2: 798.44, found: 798.3 (M+). UV-vis (in chloroform): λmax (log ε) 701 (0.49), 665 (0.41), 648 (0.17), 603 (0.09), 343 (0.25).
To an ice-cooled solution of 5 (120 mg, 150 mmol) in dichloromethane (20 mL), triethylamine (60 drops) and methane sulfonyl chloride (30 drops) were added with stirring. The mixture was allowed to warm to room temperature and stirring continued for 30 min. Then the resulting solution was washed with water and dried with anhydrous magnesium sulfate. The product was purified by column chromotography (silica gel) using chloroform as eluant to yield 6 as a black blue solid (90 mg, 68% yield).
A solution of phthalocyanine 6 (90 mg, 103 μmol) in THF (20 mL) and ethanol (6 mL) was brought to reflux. Then thiourea (60 mg, 0.79 mmol) was added. The reaction was monitored by TLC. After all of the starting material was consumed, aqueous sodium hydroxide solution (20%, 12 mL) was added. When the reaction monitored by TLC was complete, the resulting mixture was poured into a mixture of dilute hydrochloride acid and ice and extracted with dichloromethane. The organic phase was separated and dried with anhydrous magnesium sulfate. After removal of the solvent, the residue was purified by column chromatography using dichloromethane as eluant. Phthalocyanine disulfide 7 was obtained as a purple solid (36 mg, 43% yield). Data for 7: 1H NMR (300 MHz, C6D6): δ 3.64 (br, 2H, NH), -3.33 (br, 2H, NH), 0.54-1.95 (m, 70H), 2.98 (m, 4H, SCH2), 4.35 (m, 4H, OCH2), 7.00-9.40 (m, 24H, aryl). MALDI-TOF-MS: m/z calcd. for C100H106N16O2S2: 1626.81, found: 1627.1 (M+). UV-vis (in chloroform): λmax (log ε) 701 (0.37), 664 (0.54), 641 (0.52), 338 (0.60).
To a solution of phthalocyanine 9 (0.2 g, 0.26 mmol) and S-(4- iodophenyl) ethanethioate (72 mg, 0.26 mmol) in triethylamine (10 mL), dichlorobis(triphenylphosphine)palladium(Ⅱ) (9 mg, 13 μmol) and copper (Ⅰ) iodide (5 mg, 26 μmol) were added. The mixture was stirred at room temperature for 23 h. The solvent was evaporated under reduced pressure. The residue was purified by column chromatography using dichloromethane/methanol (50:1) as eluent. The product was obtained as a black solid (83 mg, 35% yield). Data for 10: 1H NMR (300 MHz, C6D6): β 1.67 (m, 27H, t-butyl), 2.42 (s, 3H, COCH3), 6.54-8.70 (m, 16H, aryl). MALDI-TOF-MS: m/z calcd. for C54H46N8OSZn: 918.28, found: 918.4 (M+). UV-vis (in chloroform): λmax (log ε) 692 (1.6), 617 (0.38), 350 (0.93).
Quartz slides were washed with chloroform and immersed into a solution of potassium hydroxide in 100 mL of deionized water and 250 mL methanol for 12 h. Then the quartz slides were washed thoroughly with deionized water and dried in a steam of nitrogen. The gold substrates were prepared by thermal evaporation of a layer of gold onto freshly cleaned quartz slides that had been precoated with a chromium adhesion layer. For UV-vis characterization, 5 nm of chromium was deposited on cleaned quartz slides followed by an 8 nm gold layer. Phthalocyanine SAMs were prepared by immersing gold substrates into phthalocyanine solutions in chloroform (6.7 × 10-5 mol/L and 2.9 × 10-5 mol/L for 7 and 10, respectively) for 24 h at room temperature. References for UV-vis measurements were prepared by immersing corresponding substrates into pure chloroform for 24 h. Then the substrates were washed thoroughly with chloroform and dried in a stream of nitrogen.
Several methods have been developed to synthesize substituted phthalocyanines. Among these methods, condensation of the corresponding phthalonitriles is the method usually referenced and appears to be the most successful one. A variety of uniformly substituted phthalocyanines have been synthesized through this method. There are also several reports with details to synthesize non-uniformly substitutedphthalocyanines [7, 8], however, it seems that the above method is the most satisfactory one at present. Two kinds of phthalonitriles bearing different substituents were used in the condensation. The product is a mixture of substituted phthalocyanines. The desired substituted phthalocyanine is then obtained after following a careful separation procedure. 4-Nitrophthalonitrile was usually used as a precursor for many other phthalonitriles through a nucleophilic substitution reaction of the nitro-group [9].
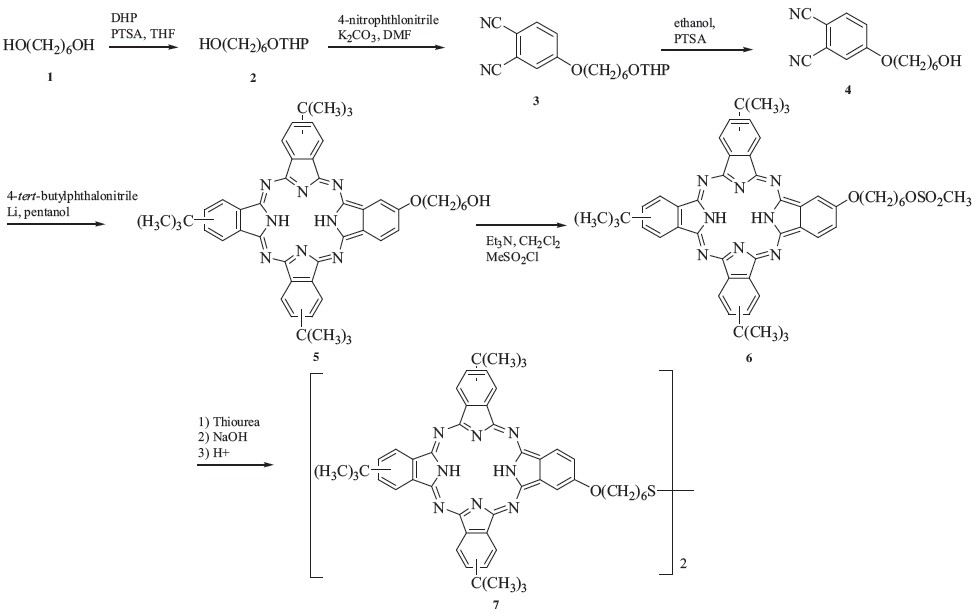
The synthetic route to phthalocyanines 7 is shown in Scheme 1. Attempts to synthesize 4-(6-hydroxyhexyloxy)phthalonitrile through the reaction of 1,6-hexanediol with 4-nitrophthalonitrile proved to be unsuccessful due to the two reactive hydroxyl groups in each diol molecule, which may react with two phthalonitrile molecules. In order to prevent this possibility, one hydroxyl group of the 1,6-hexanediol was protected by a tetrahydropyranyl group. We used a large excess of diol to react with dihydropyran, then extracted with petroleum ether to obtain 1,6-hexanediol monotetrahydropyranyl ether 2. The protected diol was then reacted with 4-nitrophthalonitrile to afford 4-(6-tetrahydropyranylhexyloxy) phthalonitrile 3. After deprotection of the hydroxy group, one of the phthalonitrile precursors 4 was obtained. Additionally, 4-tert-butylphthalonitrile was selected as another phthalonitrile to ensure that the phthalocyanines synthesized have good solubility in organic solvents. The mixed condensation of phthalonitrile 4 and 4-tert-butylphthalonitrile (1:3 ratio) was carried out in n-pentanol in the presence of lithium pentoxide to afford phthalocyanine 5 bearing a hydroxyl group. Structural isomers are possible for phthalocyanine 5 [10]. An established method was used to convert the hydroxyl phthalocyanine to the phthalocyanine disulfide [11]. The hydroxyl phthalocyanine was first mesylated, then reacted with thiourea forming an isothiuronium salt. Phthalocyanine disulfide 7 was obtained when hydrolysis of the isothiuronium salt was carried out in air. Phthalocyanine disulfide was synthesized instead of mercaptophthalocyanine. both of which can form SAM on gold substrates [4, 12]. However, the thiol can change into the disulfide in air, which makes the latter one unstable for use in an air envirnoment.

|
Download:
|
| Scheme 1.Synthesis of phthalocyanine 7. | |
{kind=link}

|
Download:
|
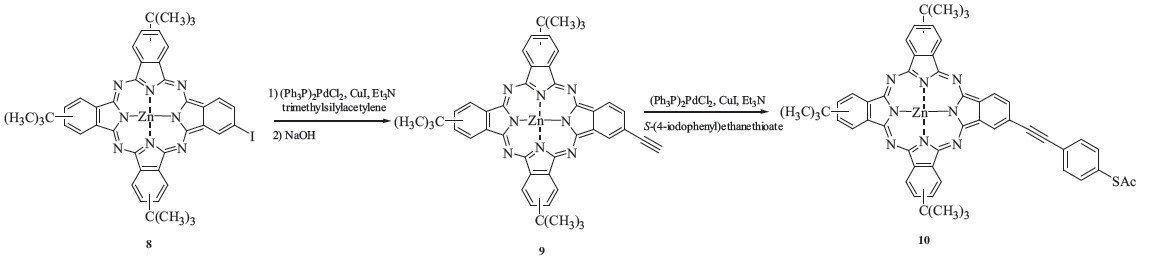
| Scheme 2.Synthesis of phthalocyanine 10. | |
{kind=link}
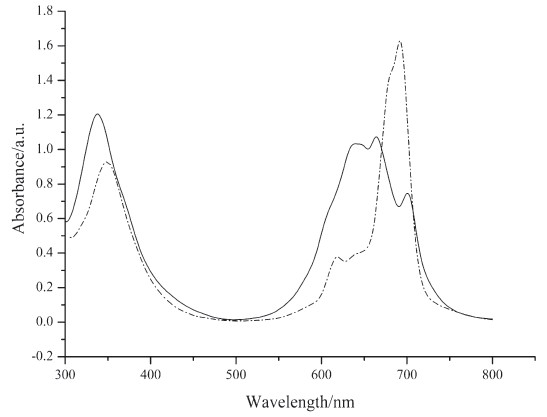
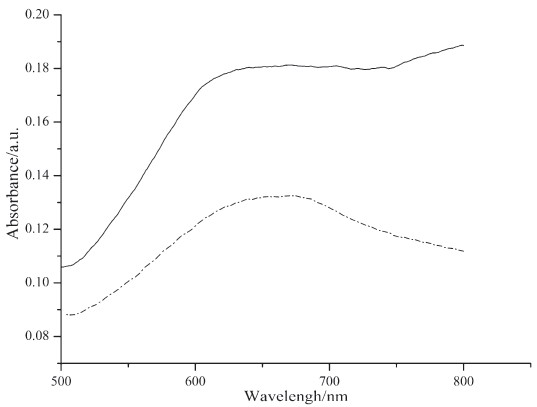
UV-vis spectra of solutions and self-assembled monolayers of phthalocyanine 7 and 10 are shown in Figs. 1 and 2. From the UV-vis spectra of these solutions (Fig. 1), we can see that the aggregation tendency of the two compounds is very high. They appear primarily as dimers as shown in theQ-band of their UV-vis spectra. UV-vis spectra of the self-assembled monolayers were measured using references prepared by immersing gold substrates on quartz slides in pure solvent. Characteristic Q-bands associated with phthalocyanines can be seen in the spectra (Fig. 2). It is obvious that the Q-bands of the SAMs are broadened and blue-shifted relative to those observed in solution (Fig. 1). Such phenomena are also observed by other authors [17, 18] and can be explained by the existence of phthalocyaninemolecules in more aggregated states in self-assembled monolayers than in solutions.

|
Download:
|
| Fig. 1.UV–vis spectra of phthalocyanine 7 (solid line) and 10 (dashed line) in chloroform. | |
{kind=link}

|
Download:
|
| Fig. 2.UV–vis spectra of self-assembled monolayers of phthalocyanine 7 (solid line) and 10 (dashed line). | |
{kind=link}
Asymmetrically substituted phthalocyanines with sulfur-containing substituents for the fabrication of self-assembled monolayers were synthesized and characterized by MALDI-TOF-MS, 1H NMR and UV-vis spectrometry. Phthalocyanine 7, bearing a disulfide group, was synthesized from phthalocyanine with a hydroxyl group via a mixed condensation of the corresponding substituted phthalonitriles. Phthalocyanine 10, bearing an acetyl protected thiol group, was synthesized through the Pd-catalysed coupling reaction of an iodophthalocyanine. Both 7 and 10 can form self-assembled monolayers on gold substrates based on the UV-vis spectra data. It was found that the Q-bands of the selfassembled monolayers were broadened and blue-shifted compared to their corresponding spectra in solutions.
This work was supported by the NSFC (Nos. 50973011 and 20702004).
| [1] | E. Gomar-Nadal, J. Puigmarti-Luis, D.B. Amabilino, Assembly of functional molecular nanostructures on surfaces, Chem. Soc. Rev. 37 (2008) 490-504. |
| [2] | E.K. Schillinger, E. Mena-Osteritz, J. Hentschel, et al., Oligothiophene versus betasheet peptide: synthesis and self-assembly of an organic semiconductor-peptide hybrid, Adv. Mater. 21 (2009) 1562-1567. |
| [3] | A. Ulman, Formation and structure of self-assembled monolayers, Chem. Rev. 96 (1996) 1533-1554. |
| [4] | X.B. Huang, Y.Q. Liu, S. Wang, et al., Synthesis and self-assembly of 2, 9,16-tri(tertbutyl)-23-(10-mercaptodecyloxy)phthalocyanine and the application of its selfassembled monolayers in organic light-emitting diodes, Chem. Eur. J. 8 (2002) 4179-4184. |
| [5] | D.J. Revell, I. Chambrier, M.J. Cook, et al., Formation and spectroscopic characterisation of self-assembled phthalocyanine monolayers, J. Mater. Chem. 10 (2000) 31-37. |
| [6] | Z.Y. Li, M. Lieberman, W. Hill, XPS and SERS study of silicon phthalocyanine monolayers: umbrella vs. octopus design strategies for formation of oriented SAMs, Langmuir 17 (2001) 4887-4894. |
| [7] | N. Kobayashi, Optically active ‘adjacent' type non-centrosymmetrically substituted phthalocyanines, Chem. Commun. (1998) 487-488. |
| [8] | K.J.M. Nolan, M. Hu, C.C. Leznoff, "Adjacent" substituted phthalocyanines, Synlett (1997) 593-594. |
| [9] | S.M. Marcuccio, P.I. Svirskaya, S. Greenberg, et al., Binuclear phthalocyanines covalently linked through 2-atom and 4-atom bridges, Can. J. Chem. 63 (1985) 3057-3069. |
| [10] | D. Wohrle, J. Gitzel, I. Okura, et al., Photoredox properties of tetra-2, 3-pyridinoporphyrazines (29H,31H-tetrapyrido[2,3-b:2',3'-g:2'',3''-i:2''',3'''-q] porphyrazine), J. Chem. Soc. Perkin Trans. 2 (1985) 1171-1178. |
| [11] | I. Chambrier, M.J. Cook, D.A. Russell, Synthesis and characterization of functionalized phthalocyanine compounds for fabrication of self-assembled monolayers, Synthesis (1995) 1283-1286. |
| [12] | J.C. Love, L.A. Estroff, J.K. Kriebel, et al., Self-assembled monolayers of thiolates on metals as a form of nanotechnology, Chem. Rev. 105 (2005) 1103-1169. |
| [13] | E.M. Maya, C. Garcia, E.M. Garcia-Frutos, et al., Synthesis of novel push-pull unsymmetrically substituted alkynyl phthalocyanines, J. Org. Chem. 65 (2000) 2733-2739. |
| [14] | M.J. Cook, M.J. Heeney, Phthalocyaninodehydroannulenes, Chem. Eur. J. 6 (2000) 3958-3967. |
| [15] | D.T. Gryko, C. Clausen, K.M. Roth, et al., Synthesis of "porphyrin-linker-thiol" molecules with diverse linkers for studies of molecular-based information storage, J. Org. Chem. 65 (2000) 7345-7355. |
| [16] | J.C. Wan, H.Q. Sun, X.B. Huang, Synthesis of a Benzenethiol-derivatized porphyrin for Self-assembly, Chin. J. Chem. 30 (2012) 1841-1844. |
| [17] | N.L. Oleinick, A.R. Antunez, M.E. Clay, et al., New phthalocyanine photosensitizers for photodynamic therapy, Photochem. Photobiol. 57 (1993) 242-247. |
| [18] | M.J. Cook, R. Hersans, J. McMurdo, et al., Self-assembled monolayers of phthalocyanine derivatives on glass and silicon, J. Mater. Chem. 6 (1996) 149-154. |