




b West China Hospital, Sichuan University, Chengdu 610041, China Union Medical College, Beijing 100050, China
Aprepitant (APT) and its prodrug,Fosaprepitant (FPT),are both potent antiemetic drugs and are well-known in the class of nonpeptide antagonists to the tachykinin neurokinin NK1 receptor [1]. They also exhibit other widespread therapeutic activities. Aprepitant has been approved by the U.S. FDA in 2003,under the name ‘‘Emend’’,for the prevention of acute and delayed chemotherapyinduced nausea and vomiting (CINV) [2]. In 2008,Fosaprepitant dimeglumine was developed as a prodrug for injection [3].
The molecular structures of Aprepitant,chemically 5-[[(2R,3S)- 2-[(1R)-1-[3,5-bis(trifluoromethyl)phenyl]-3-(4-fluoroprenyl)-4- morpholinyl]methyl]-1,2-dihydro-3H-1,2,4-triazol-3-one,and Fosaprepitant are shown in Fig. 1. Three chiral centers in their structures lead to eight isomers. Therefore,it is essential to synthesize these diastereomers involved in synthetic routes for further research.

|
Download:
|
| Fig 1 The structure of Aprepitant and Fosaprepitant. | |
{kind=link}
To the best of our knowledge [4, 5, 6],the most efficient and environmental route to synthesize Aprepitant and Fosaprepitant is shown in Scheme 1

|
Download:
|
| Scheme 1 The synthesis route of Aprepitant and Fosaprepitant. | |
{kind=link}
This route involves two typical classical steps: converting the chiral center of compound 3 under strong base to get 4,and then adding the 4-fluorobenzyl group to the amide bond to afford 5 via Grignard addition. Utilizing HPLC analysis,it is not difficult to determine that there are three major diastereomers in the final product of Aprepitant as shown in Fig. 2.

|
Download:
|
| Fig 2 Three major isomers in the synthesis route. | |
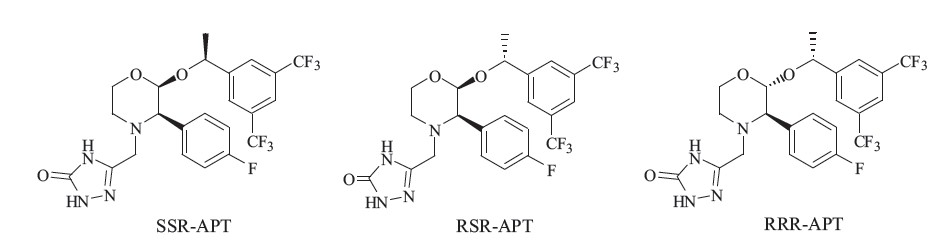{kind=link}
The formation of these three isomers was investigated. When the optical purity of the key starting material 2 is not high enough, the enantiomer SSR-APT in the final product begins to appear and is very difficult to remove. As shown in Scheme 2,the RSR-APT results from the impurity 6,which is an incompletely converted product when the chiral center is changed to yield 4. The last RRRAPT isomer results mainly from the Grignard reaction,in which hydrogenation occurred followed by the formation of the imine at room temperature in high stereo selectivity. However,we found that an increase in temperature leads to the increased RRR-APT during the reaction.

|
Download:
|
| Scheme 2 The formation of RRR-APT and RSR-APT. | |
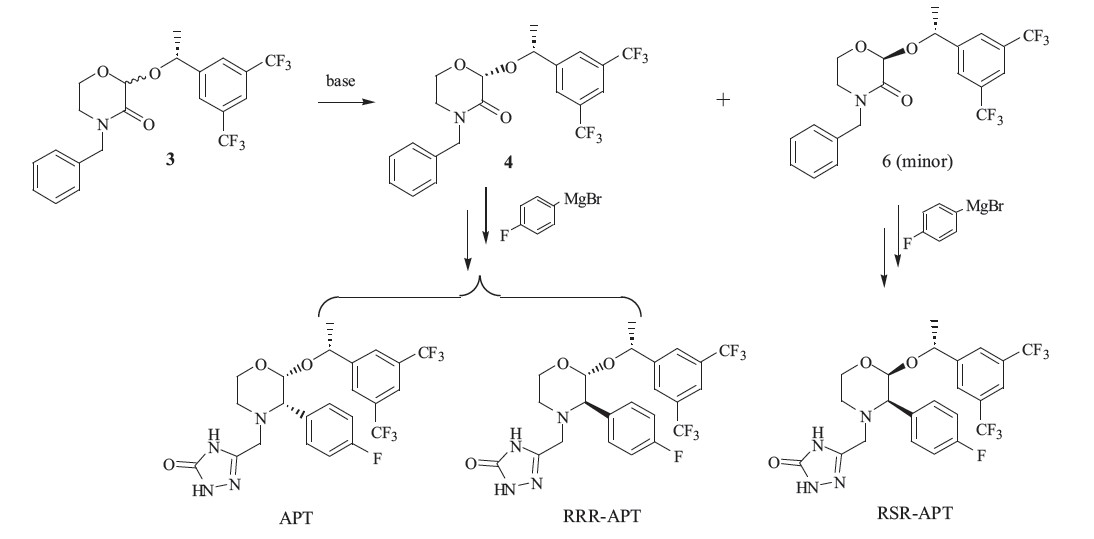{kind=link}
Those impurities in Fig. 2 are minor byproducts in the reaction, and difficult to purify by column chromatography. Gangula et al. have reported the synthesis of RRR-APT [7]. Herein,we design an efficient route to synthesize the other two impurities. As shown in Scheme 3,the coupling of 7 and 9 in acetic acid gives a mixture of 10 and its enantiomer. Upon heating this mixture in an isopropyl acetate solution of hydrogen chloride,the isomer is successfully transformed into 10 as its hydrochloric acid salt in high yield [8]. After reduction of the carboxide and coupling with 3,5-bistrifluoromethylbenzoyl chloride in one pot,ester 11 is obtained with high purity. The olefination of ester 11 with dimethyltitanocene (DMT) smoothly gives olefin 12 [9, 10, 11],and then the final reduction of 12 in Pd/C presents two enantiomers in the ratio of 3:1. Both of them are the key intermediates and easy to separate by column chromatography. By linking with side chain,we successfully synthesize the SSR-APT and RSR-APT isomers in high yield.

|
Download:
|
|
Scheme 3 Reagent and conditions: (i) 2-bromo-ethanol,Et3N,DCM,r.t.,85%; (ii) 48% HBr,DMSO,80℃,76%; (iii) AcOH,i-PrOAc,100 8C,87%; (iv) L-selectride,3,5-bistrifluoromethyl- benzoyl chloride,THF,-78℃,86%; (v) DMT,toluene,60℃,90%; (vi) 10% Pd/C,H2,TsOH,MeOH,r.t.,98% (to℃ yield for 13 and 14); (vii) K2CO3,DMF,r.t.,98%. |
|
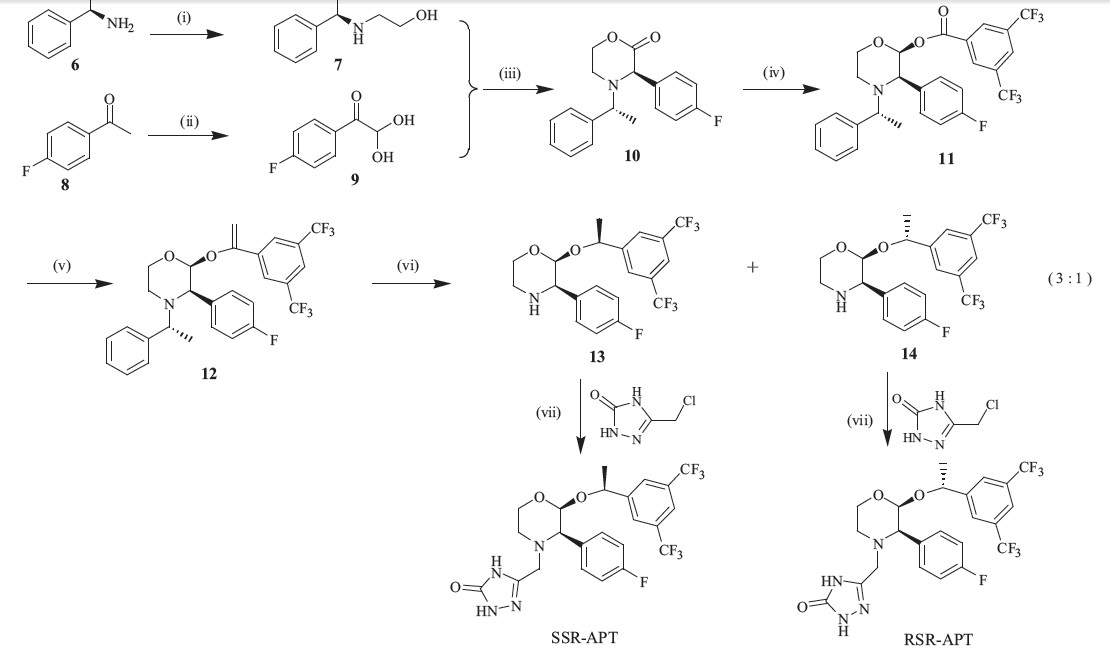{kind=link}
On the basis of the previous route,we accomplished the synthesis of the isomers of Fosaprepitant in two steps as shown in Scheme 4. Using SSR-APT as the starting material and initially reacting with tetrabenzyl pyrophosphate (TBPP) in NaHMDS gives the intermediate 15,which is unstable. After reduction in MeOH and 10% Pd/C,SSR-FPT results as a pure solid. The RSR-FPT and RRR-FPT can be obtained in the same procedure.

|
Download:
|
| Scheme 4 Reagent and conditions: (a) NaHMDS,TBPP,THF,-20℃ and (b) N-methyl-D-glucamine,10% Pd/C,5 atm H2,MeOH,r.t., 85% from SSR-APT. | |
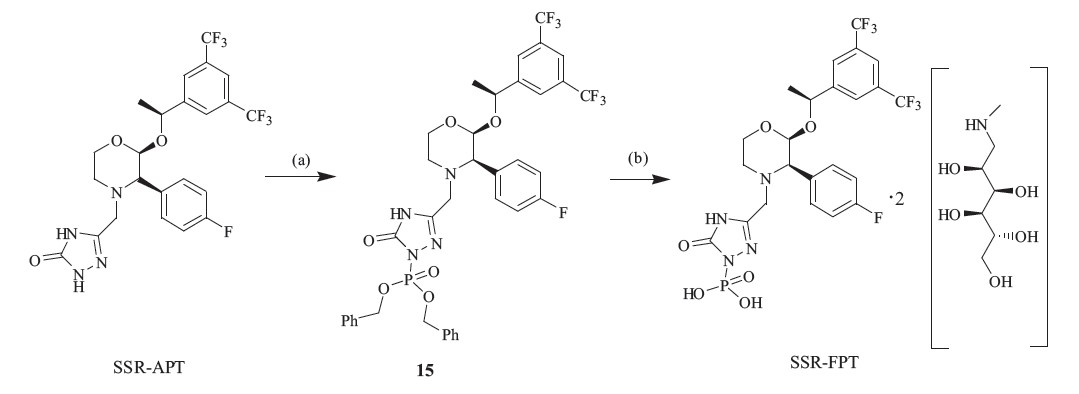{kind=link}
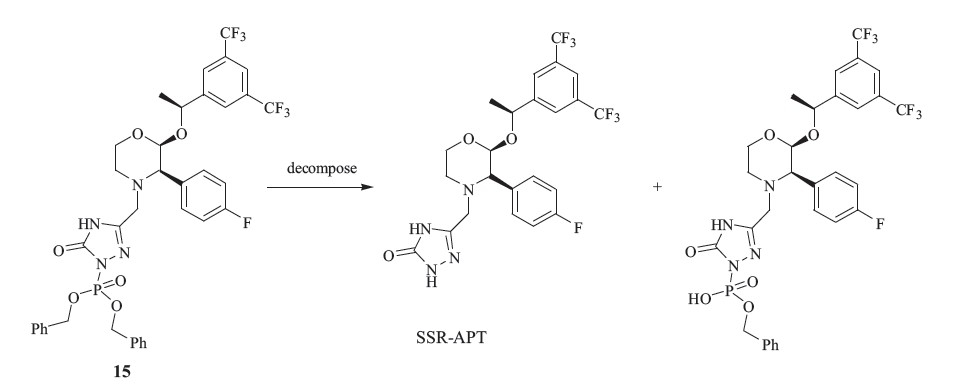
Gangula et al. reported a route to synthesize SSR-APT and RSRAPT using 1-(3,5-bis(trifluoromethyl)phenyl)ethanone as the starting material [7]. Apparently,this achiral material resulted in a complex mixture of four optical isomers,which was then directly employed to synthesize RRR-APT in five steps. The minor byproduct in the preparation of RRR-APT was purified by preparative HPLC and then used to synthesize the key intermediates 13 and 14 which were the starting materials to yield SSRAPT and RSR-APT. The RRR-APT was obtained in very low yield after a series of crystallizations and chiral resolution. The application of HPLC in the purification of the byproduct further reduced the total yield of SSR-APT and RSR-APT to less than 2%. Obviously,such an inefficient route cannot meet the demand of scaled up manufacturing. Thus,we report a directional and highperformance route as previously described. In our effort to get 9, we determined 48% HBr was essential,and that 40% HBr led to complex products. The reduction of 12 had also been studied in our work. The deprotection of α-methylbenzyl went very slowly under 10% Pd/C in the absence of acid. When 1 equiv. of ptoluenesulfonic acid was added,the reaction could be accomplished in 5 h and presented to be excellent. Compound 15 was a key intermediate in the synthesis of SSR-FPT. However,longtime storage of 15 resulted in decomposition as shown in Scheme 5. Consequently,crude 15 should be reduced immediately. The final product SSR-FPT proved to be hygroscopic and should be stored below 5 ℃.

|
Download:
|
| scheme 5 Decomposition of intermediate 15. | |
{kind=link}
In summary,we report two major isomers of Aprepitant describing their formation and the method of synthesis in high yield and the first report involving the study of the isomers of Fosaprepitant. All the target compounds were confirmed by elemental analyses,IR,NMR and MS data analysis [12]. All of the investigations are essential and important for quality control in manufacturing.
AcknowledgmentsWe would like to express our gratitude for financial support from Sichuan provincial science and technology support program (No. 2011SZ0014).
| [1] | M.M. Zhao, J.M. McNamara, G.J. Ho, et al., Practical asymmetric synthesis of Aprepitant, a potent human NK-1 receptor antagonist, via a stereoselective Lewis acid-catalyzed trans acetalization reaction, J. Org. Chem. 67 (2002) 6743-6747. |
| [2] | C. Albany, M.J. Brames, C. Fausel, et al., Randomized, double-blind, placebocontrolled, phase III cross-over study evaluating the oral neurokinin-1 antagonist Aprepitant in combination with a 5HT3 receptor antagonist and dexamethasone in patients with germ cell tumors receiving 5-day cisplatin combination chemotherapy regimens: a hoosier oncology group study, J. Clin. Oncol. 30 (2012) 3998- 4003. |
| [3] | M. Miguel, M.A. Juan, C. Rafael, et al., NK-1 receptor antagonists as antitumor drugs: a survey of the literature from 2000 to 2011, Expert Opin. Ther. Patents 22 (2012) 735-746. |
| [4] | J. Shreerang, R.A.R. Khan, N. Raji, Novel intermediates for the preparation of highly pure aprepitant or fosaprepitant, PCT Int. Appl. (2012), WO 2012146692A1. |
| [5] | N. Kolla, C.R. Elati, M. Arunagiri, et al., An alternative approach to achieve enantiopure (3S)-4-benzyl-3-(4-fluorophenyl)morpholin-2-one: a key intermediate of Aprepitant, an NK1 receptor antagonist, Org. Process Res. Dev. 11 (2007) 455-457. |
| [6] | K.M.J. Brands, J.F. Payack, J.D. Rosen, et al., Efficient synthesis of NK1 receptor antagonist aprepitant using a crystallization-induced diastereoselective transformation, J. Am. Chem. Soc. 125 (2003) 2129-2135. |
| [7] | S. Gangula, C.R. Elati, S.V. Mudunuru, et al., Synthesis of all enantiomerically pure diastereomers of Aprepitant, Syn. Commun. 40 (2010) 2254-2268. |
| [8] | M.S. Ashwood, B.C. Bishop, Chemical synthesis of 1,4-oxazin-2-ones, PCT Int. Appl. (1999), US006046325A. |
| [9] | S.R. Shenoy, K.A. Woerpel, Investigations into the role of ion pairing in reactions of heteroatom-substituted cyclic oxocarbenium ions, Org. Lett. 7 (2005) 1157-1160. |
| [10] | J.F. Payack, D.L. Hughes, F D.C. Ian, et al., Dimethyltitanocene, Org. Synth. 79 (2002) 19. |
| [11] | J.F. Payack, M.A. Huffman, D.C. David L, et al., Dimethyltitanocene: from millimole to kilomole, Org. Process Res. Dev. 8 (2004) 256-259. |
| [12] |
Physical and spectral data of the target compounds. SSR-APT: Mp: 253-255℃; [α]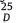 + 69:1 (c 1.0, methanol); 1H NMR (400 MHz, CD3OD): δ 7.70 (s, 1H), 7.51 (m, 2H), 7.32 (s, 2H), 7.04 (t, 2H, J = 8.7 Hz), 4.94 (q, 1H, J = 6.3 Hz), 4.35 (d, 1H, J = 2.8 Hz), 4.28 (td, 1H, J = 11.5, 2.8 Hz), 3.66 (ddd, 1H, J = 11.5, 3.3, 1.6 Hz), 3.54 (d, 1H, J = 14.3 Hz), 3.48 (d, 1H, J = 2.8 Hz), 2.88 (brd, 1H, J = 11.9 Hz), 2.86 (d, 1H, J = 14.3 Hz), 2.49 (td, 1H, J = 11.9, 3.6 Hz), 1.44 (d, 3H, J = 6.3 Hz); 13C NMR (100 MHz, CDCl3): δ 22.775, 52.302, 53.654, 60.494, 70.536, 73.746, 97.132, 116.080, 116.293, 122.367, 123.306, 126.011, 127.841, 132.401, 132.614, 133.273, 134.204, 147.074, 147.653, 158.764, 162.906, 165.351; HRMS calcd. for C23H21F7N4O3 m/z: 534.1502. Found: 557.1405 [M+Na]+. Anal. Calcd. for C23H21F7N4O3: C, 51.69; H, 3.96; F, 24.88; N, 10.48. Found: C, 51.72; H, 3.98; F, 24.85; N, 10.50. RSR-APT: [α] 25 D 37:1 (c 0.68, MeOH) (lit.[7]: [α] 25 D 38:52), Mp: 201-204℃; 1H NMR (400 MHz, CD3OD): δ 11.408 (s, 1H), 11.298 (s, 1H), 7.981 (d, 1H, J = 9.6 Hz), 7.927 (s, 2H), 7.622 (m, 2H), 7.169 (t, 2H, J = 8.8 Hz), 4.481 (d, 1H, J = 2.4), 4.732 (q, 1H, J = 6.4 Hz), 3.853 (t, 1H, J = 10.8 Hz), 3.638 (d, 1H, J = 2.4 Hz), 3.475 (d, 1H, J = 10.8 Hz), 2.905 (s, 2H), 2.850 (d, 1H, J = 13.6 Hz), 2.353 (dt, 1H, J = 8.8, J = 2.4 Hz), 1.017 (d, 3H, J = 6.4 Hz); 13C NMR (100 MHz, CDCl3): δ 21.754, 50.805, 59.570, 67.764, 73.114, 96.774, 114.603, 114.843, 121.133, 122.292, 125.000, 128.920, 129.871, 130.201, 130.524, 131.772, 133.761, 144.239, 147.952, 156.571, 160.632, 163.051; HRMS calcd. for C23H21F7N4O3 m/z: 534.1502. Found: 557.1407 [M+Na]+; Anal. Calcd. for C23H21F7N4O3: C, 51.69; H, 3.96; F, 24.88; N, 10.48. Found: C, 51.70; H, 3.95; F, 24.82; N, 10.44. SSR-FPT: Mp: 122-124℃, [α]+ 30:3. (c 1.0, methanol); 1H NMR (400 MHz, DMSO-d6): δ 7.814 (s, 1H), 7.532 (m, 2H), 7.340 (s, 2H), 7.049 (t, 2H, J = 8.8 Hz), 5.766-6.121 (brs, 12H), 4.950 (t, 1H, J = 6.4 Hz), 4.309 (d, 1H, J = 2.4 Hz), 4.091 (t, 1H, J = 11.2 Hz), 4.004 (s, 2H), 3.653 (m, 1H), 3.558 (d, 1H, J = 2.4 Hz), 3.453 (m, 10H), 3.406 (d, 1H, J = 14.0 Hz), 3.036 (m, 4H), 2.891 (d, 1H, J = 11.2 Hz), 2.661 (d, 1H, J = 14.0 Hz), 2.528 (s, 6H), 2.330 (dt, 1H, J = 8.8, 2.8 Hz), 1.373 (d, 3H, J = 6.4 Hz); 13C NMR (100 MHz, CDCl3): δ 22.288, 24.739, 33.712, 36.944, 51.157, 52.021, 59.133, 63.680, 68.480, 69.003, 70.227, 71.114, 71.459, 71.754, 114.862, 121.326, 121.986, 124.699, 126.775, 129.973, 130.303, 130.629, 131.299, 133.626, 143.204, 146.838, 156.970, 160.774, 163.199; HRMS calcd. for C37H56F7N6O16P m/z: 1004.3379. Found: 1027.3279 [M+Na]+; Anal. Calcd. for C37H56F7N6O16P: C, 44.52; H, 5.63; F, 13.25; N, 8.41; P, 3.11. RRR-FPT: Mp: 96- 98℃, [α]+ 3:8 (c 0.4, methanol); 1H NMR (400 MHz, DMSO-d6): 7.875 (s, 1H), 7.352 (m, 2H), 7.260 (m, 2H), 7.000 (t, 2H, J = 8.4 Hz), 5.023 (t, 1H, J = 6.4 Hz), 4.749 (d, 1H, J = 16.8 Hz), 4.147 (d, 1H, J = 6.4 Hz), 4.004 (s, 2H), 3.907 (d, 1H, J = 11.6 Hz), 3.744 (m, 1H), 3.453 (m, 10H), 3.218 (m, 1H), 3.036 (m, 4H), 2.622 (d, 1H, J = 6.4 Hz), 2.528 (s, 6H), 2.501 (m, 1H), 2.209 (m, 1H), 1.275 (d, 3H, J = 6.4 Hz); 13C NMR (100 MHz, CDCl3): δ 21.368, 27.191, 31.454, 39.322, 50.011, 52.071, 60.113, 62.520, 67.391, 69.053, 71.157, 71.184, 71.229, 71.714, 115.475, 123.133, 126.992, 128.700, 128.755, 129.167, 130.353, 130.721, 131.307, 138.625, 141.252, 142.310, 159.114, 161.241, 163.557; HRMS calcd. for C37H56F7N6O16P m/z: 1004.3379. Found: 1027.3259 [M+Na]+; Anal. Calcd. for C37H56F7N6O16P: C, 44.20; H, 5.59; F, 13.30; N, 8.42; P, 3.09. RSR-FPT: Mp: 142-144℃, [α]D + 69:1 (c 1.0, methanol); 1H NMR (400 MHz, CD3OD): δ 7.70 (s, 1H), 7.51 (m, 2H), 7.32 (s, 2H), 7.04 (t, 2H, J = 8.7 Hz), 4.94 (q, 1H, J = 6.3 Hz), 4.35 (d, 1H, J = 2.8 Hz), 4.28 (td, 1H, J = 11.5, 2.8 Hz), 3.66 (ddd, 1H, J = 11.5, 3.3, 1.6 Hz), 3.54 (d, 1H, J = 14.3 Hz), 3.48 (d, 1H, J = 2.8 Hz), 2.88 (brd, 1H, J = 11.9 Hz), 2.86 (d, 1H, J = 14.3 Hz), 2.49 (td, 1H, J = 11.9, 3.6 Hz), 1.44 (d, 3H, J = 6.3 Hz); 13C NMR (100 MHz, CDCl3): δ 22.775, 52.302, 53.654, 60.494, 70.536, 73.746, 97.132, 116.080, 116.293, 122.367, 123.306, 126.011, 127.841, 132.401, 132.614, 133.273, 134.204, 147.074, 147.653, 158.764, 162.906, 165.351; HRMS calcd. for C23H21F7N4O3 m/z: 534.1502. Found: 557.1405 [M+Na]+. Anal. Calcd. for C23H21F7N4O3: C, 51.69; H, 3.96; F, 24.88; N, 10.48. Found: C, 51.72; H, 3.98; F, 24.85; N, 10.50. RSR-APT: [α] 25 D 37:1 (c 0.68, MeOH) (lit.[7]: [α] 25 D 38:52), Mp: 201-204℃; 1H NMR (400 MHz, CD3OD): δ 11.408 (s, 1H), 11.298 (s, 1H), 7.981 (d, 1H, J = 9.6 Hz), 7.927 (s, 2H), 7.622 (m, 2H), 7.169 (t, 2H, J = 8.8 Hz), 4.481 (d, 1H, J = 2.4), 4.732 (q, 1H, J = 6.4 Hz), 3.853 (t, 1H, J = 10.8 Hz), 3.638 (d, 1H, J = 2.4 Hz), 3.475 (d, 1H, J = 10.8 Hz), 2.905 (s, 2H), 2.850 (d, 1H, J = 13.6 Hz), 2.353 (dt, 1H, J = 8.8, J = 2.4 Hz), 1.017 (d, 3H, J = 6.4 Hz); 13C NMR (100 MHz, CDCl3): δ 21.754, 50.805, 59.570, 67.764, 73.114, 96.774, 114.603, 114.843, 121.133, 122.292, 125.000, 128.920, 129.871, 130.201, 130.524, 131.772, 133.761, 144.239, 147.952, 156.571, 160.632, 163.051; HRMS calcd. for C23H21F7N4O3 m/z: 534.1502. Found: 557.1407 [M+Na]+; Anal. Calcd. for C23H21F7N4O3: C, 51.69; H, 3.96; F, 24.88; N, 10.48. Found: C, 51.70; H, 3.95; F, 24.82; N, 10.44. SSR-FPT: Mp: 122-124℃, [α]+ 30:3. (c 1.0, methanol); 1H NMR (400 MHz, DMSO-d6): δ 7.814 (s, 1H), 7.532 (m, 2H), 7.340 (s, 2H), 7.049 (t, 2H, J = 8.8 Hz), 5.766-6.121 (brs, 12H), 4.950 (t, 1H, J = 6.4 Hz), 4.309 (d, 1H, J = 2.4 Hz), 4.091 (t, 1H, J = 11.2 Hz), 4.004 (s, 2H), 3.653 (m, 1H), 3.558 (d, 1H, J = 2.4 Hz), 3.453 (m, 10H), 3.406 (d, 1H, J = 14.0 Hz), 3.036 (m, 4H), 2.891 (d, 1H, J = 11.2 Hz), 2.661 (d, 1H, J = 14.0 Hz), 2.528 (s, 6H), 2.330 (dt, 1H, J = 8.8, 2.8 Hz), 1.373 (d, 3H, J = 6.4 Hz); 13C NMR (100 MHz, CDCl3): δ 22.288, 24.739, 33.712, 36.944, 51.157, 52.021, 59.133, 63.680, 68.480, 69.003, 70.227, 71.114, 71.459, 71.754, 114.862, 121.326, 121.986, 124.699, 126.775, 129.973, 130.303, 130.629, 131.299, 133.626, 143.204, 146.838, 156.970, 160.774, 163.199; HRMS calcd. for C37H56F7N6O16P m/z: 1004.3379. Found: 1027.3279 [M+Na]+; Anal. Calcd. for C37H56F7N6O16P: C, 44.52; H, 5.63; F, 13.25; N, 8.41; P, 3.11. RRR-FPT: Mp: 96- 98℃, [α]+ 3:8 (c 0.4, methanol); 1H NMR (400 MHz, DMSO-d6): 7.875 (s, 1H), 7.352 (m, 2H), 7.260 (m, 2H), 7.000 (t, 2H, J = 8.4 Hz), 5.023 (t, 1H, J = 6.4 Hz), 4.749 (d, 1H, J = 16.8 Hz), 4.147 (d, 1H, J = 6.4 Hz), 4.004 (s, 2H), 3.907 (d, 1H, J = 11.6 Hz), 3.744 (m, 1H), 3.453 (m, 10H), 3.218 (m, 1H), 3.036 (m, 4H), 2.622 (d, 1H, J = 6.4 Hz), 2.528 (s, 6H), 2.501 (m, 1H), 2.209 (m, 1H), 1.275 (d, 3H, J = 6.4 Hz); 13C NMR (100 MHz, CDCl3): δ 21.368, 27.191, 31.454, 39.322, 50.011, 52.071, 60.113, 62.520, 67.391, 69.053, 71.157, 71.184, 71.229, 71.714, 115.475, 123.133, 126.992, 128.700, 128.755, 129.167, 130.353, 130.721, 131.307, 138.625, 141.252, 142.310, 159.114, 161.241, 163.557; HRMS calcd. for C37H56F7N6O16P m/z: 1004.3379. Found: 1027.3259 [M+Na]+; Anal. Calcd. for C37H56F7N6O16P: C, 44.20; H, 5.59; F, 13.30; N, 8.42; P, 3.09. RSR-FPT: Mp: 142-144℃, [α]D
|