




The Stemona alkaloids represent a group of natural products isolated from the plants named Stemonaceae. Over 120 structurally diverse compounds have been identified so far,and many of them display unique molecular architectures and biological profiles [1, 2]. As one representative member of Stemona alkaloids, stemoamide (1,Scheme 1) was isolated from Stemona tuberosa Lour,a Chinese traditional medicine that has been used for the treatment of respiratory diseases,such as asthma,bronchitis,and tuberculosis [3]. Owing to its novel chemical structure and prominent biological activities,stemoamide has stimulated extensive synthetic efforts [4]. However,although much progress has been achieved in this field,most of the synthetic routes toward stemoamide take long linear sequences and result in racemic synthesis. Therefore,the development of more efficient and asymmetric approach for the synthesis of stemoamide remains a considerable synthetic challenge.

|
Download:
|
| Scheme 1. Dyotropic rearrangement of 3,4-cis-β-lactone and synthetic strategy toward stemoamide. | |
{kind=link}
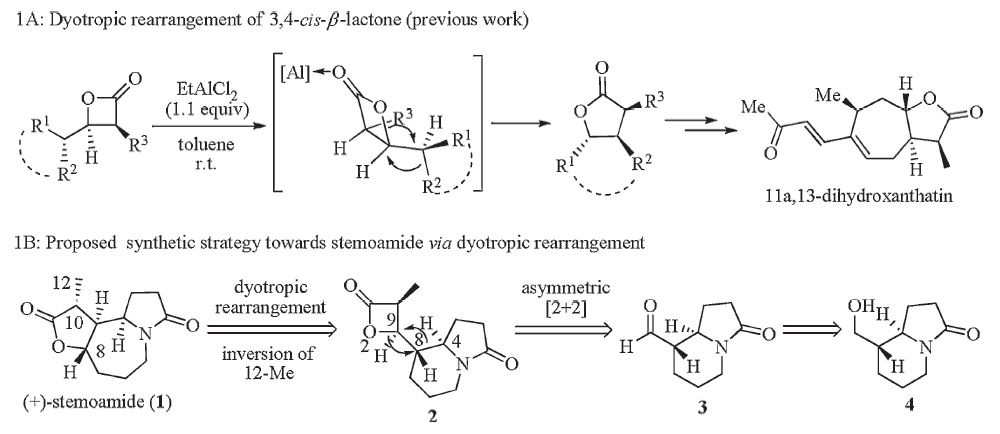
Recently,we have developed a novel approach for the syntheses of enantiopure,trisubstituted g-butyrolactones via the dyotropic rearrangement of 3,4-cis-β-lactones [5]. As the proof-of-concept case,the total syntheses of a number of xanthanolides natural products (e.g.,11α,13-dihydroxanthatin) were achieved (Scheme 1A). From the structural point of view,both xanthanolides and stemoamide feature a characteristic trisubstituted γ-butyrolactone moiety that is trans-fused with a seven-membered ring. Such structural similarity led us to assume that the 5-7-5 tricyclic core of stemoamide could also be made from the chiral β-lactone 2 through a dyotropic rearrangement,as depicted in Scheme 1B. We envisioned that the concerted nature of dyotropic rearrangement in conjunction with the rationally arranged stereochemistry of 2 (the migrating C4-C8 bond and C9-O2 bond positioned in the antiperiplanar relationship) could ensure the proposed transformation to occur in the desired manner. To validate the assumption,the asymmetric synthesis of β-lactone 2 is required,which might be achieved through an asymmetric ketene-aldehyde [2+2] cycloaddition from the aldehyde 3. In turn,3 could be derived from the alcohol 4 via oxidation. 2. Experimental
The procedure for preparation of all the intermediates in Scheme 2 and Scheme 3 are included in the Supporting information,and some selected examples (for compounds 9,4 and 2) are listed as below:

|
Download:
|
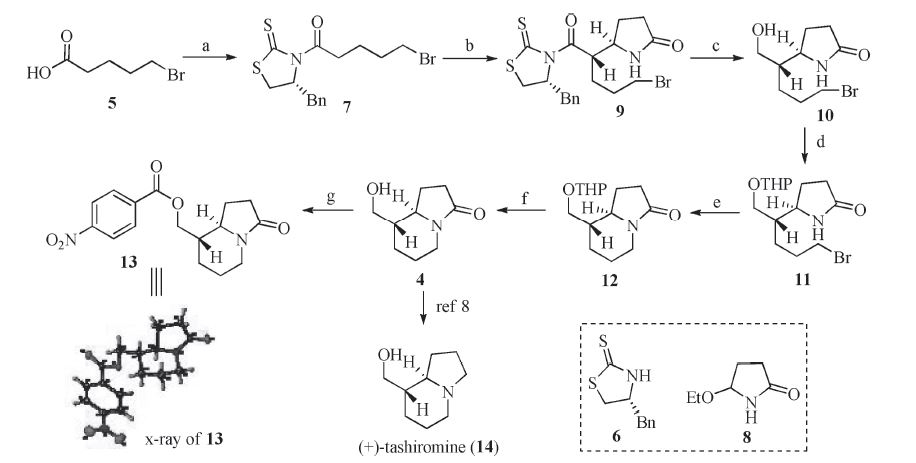
| Scheme 2. Reagents and conditions: (a) (COCl)2,DMF,DCM,r.t.,then 6,Et3N,r.t.,80%; (b) TiCl4,DIPEA,then 8,DCM -78 ℃; (c) NaBH4,MeOH,r.t.,60% for two steps; (d) 3,4-dihydro-2-pyran,p-toluenesulfonic acid pyridine salt,DCM,r.t.; (e) NaH,THF,r.t.,89% for two steps; (f) p-toluenesulfonic acid,MeOH,r.t.,90%; (g) 4-nitrobenzoyl chloride,Et3N,DCM,r.t.,80%. | |
{kind=link}

|
Download:
|
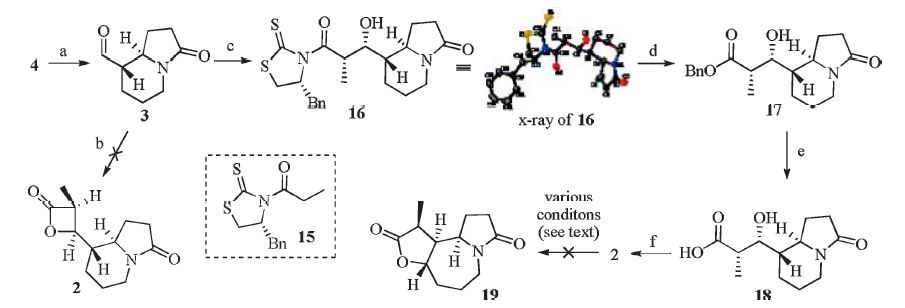
| Scheme 3. Reagents and conditions: (a) DMP,DCM,r.t.,80%; (b) TMSQ,LiClO4,propionyl chloride,DCM,-40 ℃; (c) TiCl4,DIPEA,NMP,15,DCM,-78 ℃,35%; (d) BnOH,DMAP,DCM,60%; (e) H2,Pd/C,MeOH,r.t.; (f) BOP-Cl,Et3N,DCM,r.t.,50%. | |
{kind=link}
Synthesis of compound 9: To a solution of compound 7 (50 mg, 0.14 mmol,1.0 equiv.) in DCM (2.5 mL) at 0 ℃ was added TiCl4 (0.03 mL,0.27 mmol,2.0 equiv.),and the reaction mixture was stirred for 5 min. After cooling to -30 ℃,the reaction mixture was treated with a solution of diisopropylethylamine (DIPEA) (0.026 mL,0.15 mmol,1.2 eq.) in CH2Cl2 (1 mL),and then stirred for 40 min. The reaction mixture was further cooled to 78 ℃,and a solution of compound 8 in CH2Cl2 (0.5 mL) was added. After 10 min,the reaction mixture was allowed to warm to 0 ℃ and kept stirring for 6 h before it was quenched with saturated NH4Cl. The aqueous layer was extracted with CH2Cl2 (3 × 10 mL),and the combined organic layers were washed with brine,dried over Na2SO4,filtered,and concentrated. The residue was purified by flash chromatography (DCM/EtOAc,3:1) to afford compound 9 as yellow oil (60 mg,91%). [α]D25 -216.1 (c 1.4,CH3Cl); Rf = 0.62 (silica gel,DCM/E EtOAc,3:2); IR (film,cm-1): vmax 2121,1691, 1502,1257,1165,829; 1H NMR (400 MHz,CDCl3): δ 7.26-7.34 (m, 5H),6.18 (s,1H),5.36-5.41 (m,1H),4.93 (m,1H),4.05 (dd,1H, J = 12.0,6.4 Hz),3.34-3.38 (m,3H),3.20 (d,1H,J = 12.8 Hz),3.02- 3.07 (m,1H),2.95 (d,1H,J = 12.0 Hz),2.24-2.38 (m,3H),1.98-2.06 (m,1H),1.67-1.90 (m,5H); 13C NMR (100 MHz,CDCl3): d 202.6, 177.9,175.3,136.1,129.5,129.0,127.5,68.8,55.2,46.1,37.1,32.8, 32.5,30.3,29.8,26.9,24.3; HRMS (ESI) m/z [M+H]+ calcd. for C20H25BrN2O2S2: 457.0437,found: 457.0437.
Synthesis of compound 4: A solution of compound 12 (253 mg, 1 mmol,1.0 equiv.) and p-toluenesulfonic acid (25 mg,0.1 mmol, 0.1 equiv.) in methanol (4 mL) was stirred at room temperature for 3 h. The solvent was evaporated in vacuo,and the residue was purified by flash chromatography (DCM/EtOAc,1:1) to afford compound 4 as colorless oil (150 mg,90%). [α]D25 +85.0 (c 1.3,acetone); Rf = 0.33 (silica gel,DCM/MeOH,20:1); IR (film,cm-1): vmax 3386,2920,1661,1460,1369,1050,731; 1H NMR (400 MHz, CDCl3): δ 4.13 (dd,1H,J = 12.8,4.4 Hz),3.57-3.69 (m,2H),3.24- 3.28 (m,1H),2.53-2.60 (m,1H),2.28-2.39 (m,3H),1,92-1.96 (m, 1H),1.67-1.76 (m,4H),1.29-1.43 (m,3H); 13C NMR (100 MHz, CDCl3): δ 173.9,59.2,46.1,40.0,30.6,27.1,24.6,24.1; HRMS (ESI) m/z [M+ H]+ calcd. for C9H15NO2: 170.1176,found: 170.1177.
Synthesis of compound 2: A suspension of compound 16 (20 mg,0.09 mmol,1.0 equiv.) in DCM (2 mL) was treated with Et3N (0.04 mL,0.27 mmol,3 equiv) and BOP-Cl (33 mg,0.13 mmol, 1.5 equiv.) at 23 ℃. After stirring for 2 h,the resulting reaction mixture was diluted with water (10 mL) and extracted with EtOAc (3 × 10 mL). The combined organic phases were dried and concentrated in vacuo. The residue was submitted to chromatograph (DCM/MeOH,20:1) to afford the pure product 2 as white solid (10 mg,50%). [α]D25 +22.1 (c 0.6,CH3Cl); Rf = 0.30 (silica gel, DCM/MeOH,20:1); IR (film,cm-1): vmax 2958,1821,1764,1677, 1442,1270,1144,1114,759; 1H NMR(400 MHz,CDCl3): δ 4.48 (dd, 1H,J = 6.4,4.4 Hz),4.08-4.17 (m,1H),3.85-3.88 (m,1H),3.30-3.36 (m,1H),2.58-2.64 (m,1H),2.36-2.40 (m,1H),2.22-2.33 (m,1H), 2.00-2.04 (m,1H),1.83-1.86 (m,1H),1.56-1.66 (m,5H),1.42-1.49 (m,2H),1.38 (d,3H,J = 7.6 Hz); 13C NMR (100 MHz,CDCl3): d 173.7,171.6,74.8,59.4,47.9,42.8,40.0,30.3,29.8,25.5,24.8,23.6; HRMS (ESI) m/z [M+Na]+calcd. for C12H17NO3: 224.1281,found: 224.1280. 3. Results and discussion
We initiated our study by preparing the enantiopure compound 4. Although several racemic syntheses of 4 were documented [6, 7], to date only one asymmetric synthesis of (+)-4 has been achieved, which required 11 linear steps and resulted in 18% overall yield [8]. To keep our synthesis as concise as possible,we developed a new approach to access (+)-4,as described in Scheme 2. Thus,treatment of the acid 5 with oxalyl chloride provided the corresponding acid chloride,which was coupled with thiazolidinethione 6 in the presence of Et3N to give compound 7 in 80% yield. The Lewis acidpromoted Mannich-type reaction between 7 and ethoxy-2- pyrrolidine 8 proceeded with high diastereoselectivity to afford compound 9 [9],which after treatment with NaBH4/MeOH,gave the alcohol 10 in 60% yield for two steps. Protection of the hydroxyl group with THP followed by treatment of the resulting product 11 with NaH furnished the bicyclic compound 12 in good yield. Removal of the THP group of 12 with TsOH/MeOH led to the formation of (+)-4 in an excellent yield (90%). The relative stereochemistry of 4 was unambiguously confirmed by the Xray crystallographic study of its derivative 13,while its absolute configuration was verified in the following experiments (see the Xray of 16). Compound (+)-4 could be converted into (+)- tashiromine (14) in a single step according to the literature method [8],thus our work represents a formal synthesis of (+)-14 via 7 linear steps in 30% overall yield.
With 4 in hand,we then moved forward to synthesize the chiral β-lactone 2. At the outset,the asymmetric [2+2] cycloaddition was attempted using conditions reported by Nelson [10]. Although this protocol has been successfully applied in our previous work [5],we failed to get satisfactory results under the current scenario,and only observed the recovery of starting material. To circumvent this problem,we adopted a detour strategy. Thus,the asymmetric aldol reaction between 15 and 3 afforded 16 in 35% yield [11],which was converted into the benzyl ester 17 with the action of BnOH/DMAP. Hydrolysis of the benzyl ester through hydrogenation provided the acid 18 in high yield,which after treatment with BOP-Cl/Et3N [12], formed the 3,4-cis-β-lactone 2 in 50% yield. With the precursor 2 in hand,we next turned to examine the ambitious dyotropic rearrangement to synthesize the tricyclic compound 19,which in principle,could be further converted into (+)-1 by inversion of the stereochemistry of 12-Me. However,to our disappointment, we failed to observe the desired transformation using the standard conditions (Et2AlCl,DCM,r.t.) [5]. Further efforts to improve the reaction by employing different Lewis acids (e.g. EtAlCl2,TiCl4 and MgBr2) also turned out to be fruitless. Comparing this result with the previous one [5],we assumed that the failure of the dyotropic rearrangements of 2 could be attributed to the presence of the lactam ring which may coordinate to the Lewis acid and preclude the rearrangement from occurring. 4. Conclusion
In summary,we have completed a highly efficient asymmetric formal synthesis of (+)-tashiromine. Furthermore,the total synthesis (+)-stemoamide (1) via a novel dyotropic rearrangement of 3,4-cis-β-lactone was also explored. Albeit this route failed to attain the intended objective,the experimental results shed light on the substrate limitation of the dyotropic rearrangement of 3,4- cis-b-lactone,which prompted us to devise an alternative strategy to achieve the compounds. The relevant work is in progress and will be reported in due course.
We gratefully acknowledge the financial supports from Beijing Natural Science Foundation (No. 2132037),NSFC (Nos. 21102081, 21272133),New Teachers’ Fund for Doctor Stations Ministry of Education (No. 20110002120011) and Scientific Research Foundation for the Returned Overseas Chinese Scholars,Ministry of Education (No. 20121027968).
| [1] | R.A. Pilli, D. Conceiçâo, F. Oliveira, Recent progress in the chemistry of the Stemona alkaloids, Nat. Prod. Rep. 17 (2000) 117-127. |
| [2] | R.A. Pilli, B.R. Giovanni, D. Conceiçâo, O. Ferreira de, The chemistry of Stemona alkaloids: an update, Nat. Prod. Rep. 27 (2010) 1908-1937. |
| [3] | W.H. Lin, Y. Ye, R.S. Xu, Chemical studies on new Stemona alkaloids, Ⅳ. Studies on new alkaloids from Stemona tuberosa, J. Nat. Prod. 55 (1992) 571-576. |
| [4] | K. Shishido, Recent advances in the total synthesis of xanthanolide sesquiterpenoids, Heterocycles 78 (2009) 873-889. |
| [5] | W.W. Ren, Y.C. Bian, Z.Y. Zhang, et al., Enantioselective and collective syntheses of xanthanolides involving a controllable dyotropic rearrangement of cis-b-lactones, Angew. Chem. Int. Ed. 51 (2012) 6984-6988. |
| [6] | S.P. Marsden, A.D. McElhinney, Total synthesis of the indolizidine alkaloid tashiromine, Beilstein J. Org. Chem. 4 (2008) 1-5. |
| [7] | S.H. Kim, S.I. Kim, S. Lai, et al., Titanium-Mediated cyclization of v-vinyl imides in alkaloid synthesis: isoretronecanol, trachelanthamidine, 5-epitashiromine, and tashiromine, J. Org. Chem. 64 (1999) 6771-6775. |
| [8] | D.C. Ha, S.H. Park, K.S. Choi, et al., Synthesis of (+)-tashiromine and (+)-5-epitashiromine utilizing the diastereoselective alkylation of (S)-4-arbethoxymethyl-2oxazo-lidinone, Bull. Korean Chem. Soc. 19 (1998) 728-730. |
| [9] | E. Barragán, H.F. Olivo, M. Romero-Ortega, et al., Stereoselective addition of the titanium enolate of N-acetyl (4S)-isopropyl-1,3-thiazolidine-2-thione to fivemembered N-acyl iminium ions, J. Org. Chem. 70 (2005) 4214-4217. |
| [10] | C. Zhu, X. Shen, S.G. Nelson, Cinchona alkaloid-Lewis acid catalyst systems for enantioselective ketene aldehyde cycloadditions, J. Am. Chem. Soc. 126 (2004) 5352-5353. |
| [11] | M.T. Crimmins, J. She, An improved procedure for asymmetric aldol additions with N-actyl oxazolidinones, oxazolidinethiones and thiazolidinethiones, Synlett 8 (2004) 1371-1374. |
| [12] | E.J. Corey, W.D. Li, G.A. Reichard, A new magnesium-catalyzed doubly diastereoselective anti-aldol reaction leads to a highly efficient process for the total synthesis of lactacystin in quantity, J. Am. Soc. Chem. 120 (1998) 2330-2336. |