 2019, Vol. 35
2019, Vol. 35


2. 广东省矿物物理与材料研究开发重点实验室, 中国科学院广州地球化学研究所, 广州 510640;
3. 中国科学院大学, 北京 100049
2. Guangdong Provincial Key Laboratory of Mineral Physics and Materials, Guangzhou Institute of Geochemistry, Chinese Academy of Sciences, Guangzhou 510640, China;
3. University of Chinese Academy of Sciences, Beijing 100049, China
地球环境是一个动态的物理化学系统,而该系统中可能引起各种矿物相变化的物理化学反应过程大都开始于矿物的表面或界面(Putnis, 2014)。无论是在单个矿物晶体上还是它们的各类集合体中,各种发生于矿物表面的吸附、离子交换、电子转移、沉淀、溶解、固溶体形成、水解和聚合等化学反应,或是矿物晶核发育、生长和消融,以及元素富集(成矿)与分散(环境污染)等地球化学过程,实际上主要发生在包绕于矿物颗粒体相外的各个晶面上。
自然界中,绝大多数矿物颗粒的真实表面实际上因风化、冲蚀及构造应力、剪切破碎等地质作用而由数量不等、对应于不同米勒指数的晶面包绕所组成的。其中, 既可能包含能够形成单形的低米勒指数晶面,又可能是包含高米勒指数的任意破裂面,或具较大面网密度的矿物解理面。这些矿物颗粒的实际表面由于表层原子排列截断、重构、弛豫、表面羟基化等等因素的影响,形成了不同于晶体相内部周期性结构的特殊面网,在结构上与内部面网的原子结构和配位环境截然不同,因而会表现出特殊的物理化学性质。
已有研究表明,矿物不同晶面的结构及表面物理化学性质会有很大差异(索莫杰, 1991)。由于制样条件等的限制,目前关于矿物表面物理化学性质的研究大多还是采用矿物粉晶作为研究对象,例如,传统的矿物-水界面反应性多以机械粉碎至一定粒径的矿物粉末为研究对象。对于这些经过破碎的矿物颗粒而言,无论是矿物谱学性质还是宏观表面化学反应性的研究,其结果通常只是众多矿物颗粒中无数组晶面性质的总体加和或平均。由于不同矿物各向异性的差异及其解理特性的不同,各组晶面出现的几率难以统计区分。这种以粉晶为研究对象的研究过程无疑很难获得各个晶面/解理面的准确结构及其反应性信息,导致对相关地球化学过程的认识和理解可能存在局限性。对具有极完全、完全解理的矿物而言,在机械外力的破碎过程中保留下来的低能解理面较多,因而在某种程度上能够反映实际的地球化学过程; 而对于中等、不完全、极不完全解理的矿物,在实验过程中,对样品的破碎加工等处理均可能导致被测样品形成较多具有高反应活性的高能面(断口),使得测定结果“虚高”,并高估某些重要矿物表面的反应速率。
自然界中大部分的矿物-水界面反应发生在天然矿物的稳定(低能)晶面上。从低能晶面角度研究矿物表/界面反应性可以更为准确、深入地了解矿物表面发生的地球化学反应过程。本研究以笔者团队近期开展的黄铁矿不同晶面的反应性研究为例(Zhu et al., 2018),揭示了黄铁矿{100}、{210}、{111}三个低能晶面的微观结构及其在不同湿度条件下的晶面氧化行为的差异,阐明了晶面结构制约黄铁矿氧化的机理,对理解复杂的地球化学过程与机制具有极其重要的意义。
1 “粉晶研究”在处理黄铁矿氧化问题上的局限性黄铁矿氧化过程在环境科学和地球化学领域颇受关注,因为它不仅在形成酸性矿山废水(Acid mine drainage, AMD)(Ouyang et al., 2015; Blasco et al., 2016)和湖底沉积物氧化酸化方面具有重要的环境意义,而且它也是全球硫循环中的一个重要环节(Hurtgen, 2012; Rickard et al., 2017)。因此,前人在这方面曾做过大量研究,其中,有关黄铁矿氧化研究的核心问题是氧化速率和氧化机理。
表 1列出了文献中报道的黄铁矿在潮湿空气中氧化的速率。由表可以看出,前人对黄铁矿氧化速率的测定值大相径庭,不同文献中报道的反应速率跨度超过两个数量级。如此宽泛的反应速率,在相应的地球化学进程或环境评估过程中,很难做出准确判断和估算。
|
|
表 1 文献中报道的黄铁矿在潮湿空气中氧化的速率 Table 1 Oxidation rates of pyrite surfaces in humid air reported in the literatures |
黄铁矿的氧化过程比较复杂,既涉及Fe元素的氧化又涉及S元素的氧化,但由于S元素的价态多变,增大了人们对黄铁矿氧化机理认识的难度。目前,关于黄铁矿的氧化过程可大体由如下两个总反应式进行描述(Nordstrom and Southam, 1997):
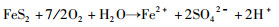
|
(1) |
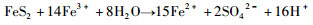
|
(2) |
尽管已有大量研究专注于黄铁矿的氧化机理(Chandra and Gerson, 2010),其中包括有精心设计的动力学实验(Jerz and Rimstidt, 2004; Osborne et al., 2010; Sun et al., 2015)和一些原位测试(Nesbitt and Muir, 1994; Guevremont et al., 1998; Zhao et al., 2010; Chandra and Gerson, 2011),但黄铁矿的氧化机理本质仍不清楚。比如,Morth and Smith (1966)认为,黄铁矿氧化的速控步(rate-limiting step)为由氧气解离所形成的活化配合物的分解作用,他们仅将水作为反应媒介而不是一种反应物。采用XPS、UPS、STM以及蒙特·卡洛(Monte Carlo)模拟方法,Eggleston et al. (1996)认为Fe2+和Fe3+之间的循环是黄铁矿氧化过程中的一个重要特征。Jerz and Rimstidt (2004)通过测量含氧气和黄铁矿的反应器与参比反应器的气压差,认为空气中氧分压是控制黄铁矿氧化速率的主要因素。
为了厘清黄铁矿的氧化过程,Rimstidt and Vaughan (2003)详细描述了黄铁矿氧化过程的可能反应。他们将黄铁矿的氧化看成发生在Fe和S位上的电化学反应,Fe位为阴极,S位为阳极,且总反应的速控步由阴极Fe位的反应所控制。除此之外,近期的DFT计算表明,黄铁矿氧化的速控步涉及两种表面羟基的形成,也就是说速控步与水有关(Dos Santos et al., 2016)。由此可见,已有的对黄铁矿氧化机理的解释众说纷纭,且部分观点是相互对立或矛盾的,特别是对于水在黄铁矿氧化过程中所起的作用还不明确。导致这一现状的一个重要原因是很多研究结果只是基于黄铁矿的粉晶实验,或仅仅是黄铁矿{100}一种晶面的理论计算与实验数据,没有考虑到黄铁矿晶面反应的差异性问题。
2 黄铁矿结晶习性与典型低能晶面的稳定性黄铁矿广泛分布于三大岩类中,常以各种单形晶或聚形晶出现。砂川一郎(Sunagawa, 1957)通过对日本花冈矿山的66种黄铁矿单形的测量发现,黄铁矿的三种主要单形出现频率高低顺序依次为立方体{100}、五角十二面体{210}、八面体{111}。陈光远等(1987)通过对胶东三县四个金矿区2495颗黄铁矿晶体中单形出现频率统计也发现类似规律,即在单形晶体中,黄铁矿均由{100}、{210}、{111}单形组成,相应的出现频率比值为224︰42.8︰1,且94.74%的聚形晶均是由这三种单形组合而成。可见,黄铁矿在自然界的结晶习性为立方体{100}、八面体{111}、五角十二面体{210}三种单形晶及由这三种单形构成的不同聚形晶(图 1)。

|
图 1 黄铁矿的常见结晶习性(据Murowchick, 1985) Fig. 1 Common pyrite crystal habits(after Murowchick, 1985) |
黄铁矿在自然界出现的单形晶频率与前人理论研究(Hung et al., 2002a, b; Alfonso, 2010)获得的黄铁矿最稳定的三种晶面是一致的,即{100}、{210}、{111}。关于黄铁矿这三组晶面稳定性的研究已有大量的文献报道。文献中黄铁矿不同晶面的表面能数据详见表 2。综合这些数据来看,文献已报道的四个晶面的稳定性顺序为:{100}>{111}>{210}>{110}。这一稳定性顺序均是在分析具有化学计量比的表面上进行的,难以反映实际情况下真实存在的表面结构稳定性。为解决这一问题,Alfonso(2010)考察了这些晶面所有可能的原子终止面,并计算了这些面在不同的硫分压条件下的表面能(图 2)。不难发现,在不同硫环境下发育的最稳定黄铁矿晶面为{100}-S、{111}-3S和{210}-2S′。除{100}-S为符合FeS2化学计量比的黄铁矿表面外,其余两个面均是富硫表面。这与在地质体中和模拟实验中观察到的{111}和{210}常产出于硫逸度较高的环境一致(Murowchick, 1985; 蔡元吉, 1990)。因此,本研究以黄铁矿{100}、{210}、{111}面为研究对象,旨在考察不同晶面的氧化反应性异同。
|
|
表 2 已报道的黄铁矿不同晶面的表面能(J/m2) Table 2 Summary data about the reported surface energy of different crystal faces of pyrites (J/m2) |

|
图 2 计算获得的黄铁矿不同晶面的表面能随硫化学势的变化趋势(据Alfonso, 2010) 图上方的横坐标表示在T=300、600和900K条件下的S2蒸汽压 Fig. 2 Calculated surface free energies of various pyrite surfaces as functions of the change in S chemical potential (after Alfonso, 2010) Corresponding S2 pressure axes at T=300, 600, and 900K are included |
本文以天然立方体{100}、八面体{111}、五角十二面体{210}黄铁矿单晶作为研究对象。立方体和八面体黄铁矿单晶样品产自西班牙Navajun;五角十二面体黄铁矿单晶样品产自中国湖南。这些天然黄铁矿晶体的电子探针数据(参见表 3)表明,样品中均含有少量As、Co、Ni、Au等杂质元素,但总含量均低于0.5%。根据本研究的最终结果发现,晶面结构在黄铁矿表面氧化反应中起主导作用,因此本研究将杂质元素对黄铁矿表面氧化反应的影响作为次要因素忽略,主要考察晶面结构的影响。
|
|
表 3 本研究所用的黄铁矿单晶样品的元素组成(wt%) Table 3 Elemental compositions (wt%) of the single pyrite crystals used in this study |
氧化实验所使用的样品是从上述天然黄铁矿单晶中定向切割加工而来。这些天然单晶采用金刚石低速锯将具有镜面级平整的晶面部分切割成块体。切割后的样品表面采用扫描电子显微镜(SEM)进行表面形态确认,以确保实验所用的样品表面不存在微裂纹等可能影响实验结果的因素。不同黄铁矿晶面的晶体学取向采用背散射电子衍射(EBSD)进行确认。然后用这些包含{100}、{111}、{210}面的黄铁矿块体在湿度控制的空气气氛条件下进行氧化实验,具体实验过程可参考文献(Zhu et al., 2018)。
采用X射线光电子能谱(XPS)作为黄铁矿表面氧化过程中氧化产物的检测手段,根据XPS谱中氧化产物和未被氧化的物种的相对含量可以半定量地表征这些黄铁矿不同晶面的氧化程度。图 3中给出了黄铁矿表面上氧化产物和未被氧化的物种之间的比值(Coxidized/Cunoxidized)与氧化时间(t)的函数关系,总体呈现出明显的正相关关系。根据图 3的横、纵坐标定义可知,图 3中的斜率k(hkl)代表了黄铁矿的平均氧化速率。可以看出,在相同的湿度条件下,不同黄铁矿晶面的S 2p和Fe 2p3/2谱学分析结果均表现出相同氧化速率顺序,即在相对低的湿度条件(47rh%)下,k{210}>k{111}>k{100};而在相对高的湿度条件(98rh%)下,k{111}>k{210}>k{100}。

|
图 3 黄铁矿{100}、{111}和{210}面在不同相对湿度的空气中氧化过程中的S 2p和Fe 2p3/2谱中被氧化与未被氧化的物种的比值(Coxidized/Cunoxidized)与氧化时间(t)的相关性曲线 (a、b)为相对湿度47rh%;(c、d)为相对湿度98rh% Fig. 3 Plots of the concentration ratios of oxidized species to unoxidized species (Coxidized/Cunoxidized) vs. oxidation time (t) from S 2p and Fe 2p3/2 XPS spectra of pyrite {100}, {111}, and {210} surfaces oxidized in various relative humidities (a, b) denote the relative humidity of 47rh%; while (c, d) denote the relative humidity of 98rh% |
更进一步地,我们用XPS数据来定量表征黄铁矿的氧化速率。根据XPS的分析原理,XPS的样品检测深度(d)与起飞角(θ)存在如下关系:
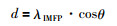
|
(3) |
式中,λIMFP为非弹性平均自由程。由于本研究采用的起飞角为0°,所以d=λIMFP。对硫而言,λIMFP近似为2.67nm(Powell et al., 2005),即d近似等于2.67nm。假设黄铁矿表面被氧化的产物的相对含量与氧化深度(x)呈正比关系,则可以获得下式:
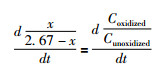
|
(4) |
式中,t为氧化时间,单位为天;k为黄铁矿晶面上被氧化与未被氧化的物种的含量比值与时间t之间线性关系的斜率,即图 3中的斜率。通过数学变换,公式2可以转化为氧化深度速率(rdepth,单位为nm/天):
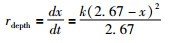
|
(5) |
从式5可知,深度速率随氧化深度的增加而减小,且当x达到XPS检测的最大深度时,深度速率则会变为零。基于图 3中最大的被氧化与未被氧化的物种的含量比Coxidized/Cunoxidized,可以估算得本研究实验的最大氧化深度约为1.19nm。因此,式5所示的速率公式仅适用于氧化深度小于1.19nm的情形。如果我们只考虑氧化反应起始阶段,即x=0nm的时候,那么,我们便可以得到如式5所示的简化初始速率公式:

|
(6) |
基于黄铁矿密度(5.0g/cm3)和黄铁矿氧化的化学计量系数,可以将深度速率(nm/天)转化为耗氧的摩尔速率(mol O2/m2s)。
图 4中标识出了本研究获得的黄铁矿{100}、{111}、{210}晶面的初始氧化速率与文献中已报道的氧化速率的对比。可以看出,前人报道的黄铁矿在潮湿空气中的氧化速率数据在10-8.7~10-5.7mol O2/m2s的范围内,而本研究获得的黄铁矿不同晶面的速率范围为10-9~10-7mol O2/m2s。根据黄铁矿不同晶面在自然界的产出频率,可以估算得黄铁矿三种晶面的加权速率为9.84×10-9~3.08×10-8 mol O2/m2s。通过对比分析可以发现,(1)前人报道的大部分黄铁矿在空气中的氧化速率均在本研究报道的晶面的氧化速率范围内;(2)部分报道(Hammack and Watzlaf, 1990)的速率远大于本研究获得的最大速率。作为一个无解理且自形程度高的矿物,自然界中黄铁矿矿物晶体主要是由{100}、{210}、{111}等相对稳定的低能晶面单形或它们之间的聚形组成,所以黄铁矿粉体的氧化速率就可以看成是黄铁矿这些低能面的统计平均数。需要指出的是,前人的数据绝大多数是基于实验室人工粉碎研磨的黄铁矿粉末而获得,这些研磨的矿物粉末通常是由不规则的微米级黄铁矿颗粒组成,颗粒表面通常主要都是一些新鲜破裂面。在这些人为形成的破裂断面中,不仅包含有低能面,也包含有一些高能面,这就有可能导致使得所测定的氧化速率偏高。

|
图 4 采用晶面研究和前人采用粉末研究获得的黄铁矿氧化速率对比 Fig. 4 Comparison of oxidation rates of pyrite observed from crystal faces in this study and from crystal powder in previous study |
根据本研究的结果可知,在具有相同米勒指数的黄铁矿晶面上,相对湿度越大其氧化程度越高;而在相同的湿度条件下,{210}和{111}晶面的氧化速率是{100}晶面的10至100倍,这与前人对黄铁矿{100}和{111}晶面的氧化速率的研究结果一致(Elsetinow et al., 2000; Guevremont et al., 1998)。然而,在相对湿度较高的条件下,{111}晶面的氧化活性比{210}晶面更高,这是前人的研究并未曾发现的。
黄铁矿在空气中的氧化主要涉及两种反应物,即O2和H2O。假设黄铁矿的氧化反应始于这两种反应物在黄铁矿晶面上的吸附,通过对比DFT计算获得的吸附能与氧化速率不难发现,H2O在黄铁矿不同晶面的吸附能绝对值大小顺序与在相对湿度较低条件下的氧化反应速率顺序一致,即{210}(-103kJ/mol)>{111}(-88.91kJ/mol)>{100}(-35.31kJ/mol);O2在黄铁矿不同晶面的吸附能绝对值大小顺序与在相对湿度较高条件下的氧化反应速率顺序一致,即{111}(-573.50kJ/mol)>{210}(-469.15kJ/mol)>{100}(-418.19kJ/mol)。
尽管我们发现了O2和H2O在黄铁矿表面的吸附能与其反应速率有一定的关联,但仍然很难获知黄铁矿氧化反应的速控步。精确的速控步需要对整个反应途径、过渡态与活化能的详细分析,因此,本研究对于速控步相关的讨论均是基于前人在黄铁矿{100}晶面获得的有限信息(Sit et al., 2012; Dos Santos et al., 2016)进行的。在上述文献中提出的反应路径中,H2O在黄铁矿{100}晶面发生伴随电子转移的解离反应的活化能(85.77~94.98kJ/mol)比O2在黄铁矿{100}晶面发生解离的活化能(25.52kJ/mol)更高。从这一角度上讲,伴随有电子转移的H2O解离可以认为是黄铁矿{100}晶面氧化的速控步。换言之,由黄铁矿表面结构本身性质导致的表面氧化反应性顺序应为{210}>{111}>{100}。
黄铁矿氧化后,在表面会形成硫酸盐等氧化产物的含水覆盖层(Jerz and Rimstidt, 2004)。因此,在形成这些氧化产物覆盖层后,氧气就需要通过扩散的方式才能到达黄铁矿表面参与反应。在相对低湿度条件下,氧化速率慢,氧化产物厚度较小。氧气在{210}和{111}晶面的扩散速率差别不大,因此,反应主要受黄铁矿表面结构控制。根据黄铁矿不同晶面的电子结构差异,这种受表面结构控制的反应速率应该是{210}大于{111}。与之对应,在相对高湿度条件下,氧化速率快,氧化产物厚度增大。因为{111}面对氧气分子的吸附能更低,所以氧气在{111}表面的氧化产物覆盖层的扩散速率比{210}更大,因此,反应主要受氧气分子在介质中的传质控制,所以在相对高湿度条件下获得了{111}的反应速率比{210}更高。综上所述,黄铁矿在低相对湿度和高相对湿度的氧化速率不同是由表面反应类型所决定的,前者受表面结构控制,而后者受传质控制。
5 黄铁矿在空气中氧化的电子转移途径根据上述结果与讨论,黄铁矿的不同晶面在空气中的氧化反应可以用前人的DFT计算(Dos Santos et al., 2016)给出的两类反应进行解释。Ⅰ型反应主要涉及黄铁矿表面的Fe(Ⅱ)被O2氧化形成Fe(Ⅲ)。这类反应的活化能较低,且需要消耗吸附在黄铁矿表面的水(Fe-OH2)。Ⅱ型反应为涉及S=O或S-OH形成的反应。这一反应过程相对Ⅰ型反应具有更高的活化能,同时会将Ⅰ型反应生成的Fe(Ⅲ)还原成Fe(Ⅱ)。可见,Fe(Ⅱ)与Fe(Ⅲ)的循环是黄铁矿氧化反应的主要特征(Eggleston et al., 1996)。一个典型的Ⅰ型和Ⅱ型反应可以分别表示为:
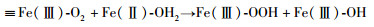
|
(7) |
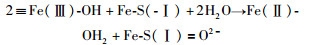
|
(8) |
这两类反应均可以看成与上面讨论的速控步一致。由于Ⅱ型反应的活化能更高,所以Ⅱ型反应可以进一步被认为速控步。Ⅱ型反应不仅涉及H2O的消耗,且H2O同时作为中间体将电子由表面S原子转移至表面Fe原子,因此在只考虑电子转移的情况下,H2O可以被认为是这一电子转移反应的催化剂。
尽管可以用水作为催化剂的电子转移过程来解释这种因黄铁矿晶面结构不同而导致其氧化反应能力不同的现象,但上面所讨论的反应途径仅仅涉及形成S=O/S-O,且这一过程的O均是来自于H2O,与前人的同位素标记实验结果不符(Balci et al., 2007; Kohl and Bao, 2011)。这些同位素标记实验表明,黄铁矿氧化形成的硫酸盐中有12.5%~25%的O来自于O2。这就意味着O2也会参与后续的S=O的进一步氧化过程。在黄铁矿的氧化过程中,除了硫酸根外,还能检测到亚硫酸根和硫代硫酸根等。由于亚硫酸根和硫代硫酸根在空气中很容易被O2氧化成硫酸根,因此,可以认为最终氧化产物硫酸根中来自于O2的O发生在这一阶段。根据Rimstidt and Vaughan (2003)提出的反应方程式,在黄铁矿表面形成S(-Ⅰ)=O/S(0)-OH后可能发生如下反应:
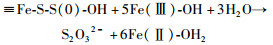
|
(9) |
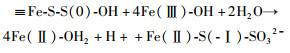
|
(10) |
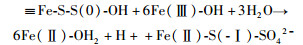
|
(11) |

|
(12) |
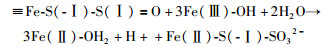
|
(13) |

|
(14) |
上面的这些反应式可以归属于Ⅱ型反应,形成的硫酸盐中的氧均来自于H2O。形成的这些硫-氧物种均为内层的配合结构。这些内层形成的S2O32-和SO32-可以被空气中O2氧化形成硫酸根,然后脱附形成外层的硫酸根,可以表示为:
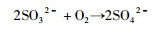
|
(15) |

|
(16) |
由这两个反应式可以计算得这两个步骤中形成的硫酸根中的O有25%至62.5%来自于O2。由于这两个反应是直接被O2氧化的,因此称之为Ⅲ型反应。而最终形成的硫酸盐既有来自于Ⅱ型反应的,也有来自于Ⅲ型反应的,因此最终形成的硫酸盐中的氧的组成应该是Ⅱ型与Ⅲ型反应的加权平均的结果。这就解释了前人的同位素标记实验中获得的硫酸盐中12.5%~25%的O来自于O2。
基于上述这些讨论,本研究将黄铁矿表面的氧化反应电子转移途径归纳成如下四步(参见图 5):

|
图 5 黄铁矿在空气中氧化反应的电子转移途径 Fig. 5 Summarized electron transfer routes of pyrite oxidation in air |
(1) 表面的Fe(Ⅱ)被O2氧化成Fe(Ⅲ)-OH;
(2) 表面S(-Ⅰ)被步骤(1)中形成的Fe(Ⅲ)-OH氧化形成S(0)-OH和S(Ⅰ)=O;
(3) S(0)-OH和S(Ⅰ)=O被步骤(1)中形成的Fe(Ⅲ)-OH氧化形成S2O32-、SO32-或SO42-;
(4) 步骤(3)中形成的S2O32-和SO32-直接被O2氧化形成SO42-。
6 结语本研究通过对具有不同表面结构的黄铁矿典型低指数晶面氧化行为的研究,从晶面结构制约其表面反应性的角度出发,以晶面各向异性的视角重新审视了黄铁矿氧化的相关问题。从这一新的视角出发,发现黄铁矿氧化速率和氧化机理研究中传统“粉末研究”实验手段有明显局限性,明确了各晶面较大的反应性差异是导致黄铁矿氧化速率宽范围的一个重要因素,约束收窄了黄铁矿氧化速率范围。水在黄铁矿的氧化过程中同时扮演着传递电子的催化剂和反应物的角色,也是黄铁矿氧化反应速控步的核心物质,以此为基础我们提出了全新的黄铁矿氧化过程全局电子转移途径。
以上研究获得的对黄铁矿不同晶面可导致的晶面反应能力截然不同的全新认识,启示我们能够而且应该将传统的表面矿物学工作推向更为精准、细致的晶面矿物学研究上来。这一从晶面结构和反应性的角度考察发生在矿物表/界面的地球化学反应行为与过程的研究方法可为构建更为精确的地球化学模型提供理论支撑。
要实现从传统的矿物粉晶表面反应性到更为精确的矿物晶面反应性的认识转变还需要开展更为广泛和深入的工作。在未来的相关矿物表/界面物理化学研究中应更加注意理论与实践并重;在深入探究由矿物晶体各向异性导致的晶面结构差异性在制约其表面反应性的深层次机理的同时,也需要查明这种由结构异同驱使的反应性差异在各类地球化学具体过程中所扮演的角色。
谨以此文献给叶大年先生八十华诞!
Alfonso DR. 2010. Computational investigation of FeS2 Surfaces and prediction of effects of sulfur environment on stabilities. The Journal of Physical Chemistry C, 114(19): 8971-8980. DOI:10.1021/jp100578n |
Balci N, Shanks Ⅲ WC, Mayer B and Mandernack KW. 2007. Oxygen and sulfur isotope systematics of sulfate produced by bacterial and abiotic oxidation of pyrite. Geochimica et Cosmochimica Acta, 71(15): 3796-3811. DOI:10.1016/j.gca.2007.04.017 |
Blasco M, Gázquez MJ, Pérez-Moreno SM, Grande JA, Valente T, Santisteban M, de la Torre ML and Bolívar JP. 2016. Polonium behaviour in reservoirs potentially affected by acid mine drainage (AMD) in the Iberian Pyrite Belt (SW of Spain). Journal of Environmental Radioactivity, 152: 60-69. DOI:10.1016/j.jenvrad.2015.11.008 |
Cai J and Philpott MR. 2004. Electronic structure of bulk and (0 0 1) surface layers of pyrite FeS2. Computational Materials Science, 30(3-4): 358-363. DOI:10.1016/j.commatsci.2004.02.026 |
Cai YJ. 1990. Experimental study of crystal forms of pyrite. Bull. Nanjing Inst. Geol. M. R., Chinese Acad. Geol. Sci., 11(2): 33-45. |
Chandra AP and Gerson AR. 2010. The mechanisms of pyrite oxidation and leaching:A fundamental perspective. Surface Science Reports, 65(9): 293-315. DOI:10.1016/j.surfrep.2010.08.003 |
Chandra AP and Gerson AR. 2011. Pyrite (FeS2) oxidation:A sub-micron synchrotron investigation of the initial steps. Geochimica et Cosmochimica Acta, 75(20): 6239-6254. DOI:10.1016/j.gca.2011.08.005 |
Chen GY, Sun DS, Zhang L, Zang WS, Wang J and Lu AH. 1987. Morphogenesis of pyrite. Geoscience, 1(1): 60-76. |
de Leeuw NH, Parker SC, Sithole HM and Ngoepe PE. 2000. Modeling the surface structure and reactivity of pyrite:Introducing a potential model for FeS2. The Journal of Physical Chemistry B, 104(33): 7969-7976. DOI:10.1021/jp0009498 |
Dos Santos EC, de Mendonça Silva JC and Duarte HA. 2016. Pyrite oxidation mechanism by oxygen in aqueous medium. The Journal of Physical Chemistry C, 120(5): 2760-2768. DOI:10.1021/acs.jpcc.5b10949 |
Eggleston CM, Ehrhardt JJ and Stumm W. 1996. Surface structural controls on pyrite oxidation kinetics:An XPS-UPS, STM, and modeling study. American Mineralogist, 81(9-10): 1036-1056. DOI:10.2138/am-1996-9-1002 |
Elsetinow AR, Guevremont JM, Strongin DR, Schoonen MAA and Strongin M. 2000. Oxidation of {100} and {111} surfaces of pyrite:Effects of preparation method. American Mineralogist, 85(3-4): 623-626. DOI:10.2138/am-2000-0428 |
Guevremont JM, Elseinow AR, Strongin DR, Bebie J and Schoonen MAA. 1998. Structure sensitivity of pyrite oxidation:Comparison of the (100) and (111) planes. American Mineralogist, 83(11-12): 1353-1356. |
Hammack RW and Watzlaf GR. 1990. The effect of oxygen on pyrite oxidation. In: Proceedings of 1990 Mining and Reclamation Conference and Exhibition. Charleston:West Virginia University: 257-264.
|
Hung A, Muscat J, Yarovsky I and Russo SP. 2002a. Density-functional theory studies of pyrite FeS2 (111) and (210) surfaces. Surface Science, 520(1-2): 111-119. DOI:10.1016/S0039-6028(02)02294-X |
Hung A, Muscat J, Yarovsky I and Russo SP. 2002b. Density-functional theory studies of pyrite FeS2 (100) and (110) surfaces. Surface Science, 513(3): 511-524. DOI:10.1016/S0039-6028(02)01849-6 |
Hurtgen MT. 2012. The marine sulfur cycle, revisited. Science, 337(6092): 305-306. DOI:10.1126/science.1225461 |
Jerz JK and Rimstidt JD. 2004. Pyrite oxidation in moist air. Geochimica et Cosmochimica Acta, 68(4): 701-714. DOI:10.1016/S0016-7037(03)00499-X |
Kohl I and Bao HM. 2011. Triple-oxygen-isotope determination of molecular oxygen incorporation in sulfate produced during abiotic pyrite oxidation (pH=2~11). Geochimica et Cosmochimica Acta, 75(7): 1785-1798. DOI:10.1016/j.gca.2011.01.003 |
Morth AH and Smith EE. 1966. Kinetics of the sulfide-to-sulfate reaction. Am. Chem. Soc. Div. Fuel Chem., 10: 83-92. |
Murowchick JB. 1985. The formation and growth of pyrite, marcasite, and cubic ferrous sulfide. The Pennsylvania State University
|
Nesbitt HW and Muir IJ. 1994. X-ray photoelectron spectroscopic study of a pristine pyrite surface reacted with water vapour and air. Geochimica et Cosmochimica Acta, 58(21): 4667-4679. DOI:10.1016/0016-7037(94)90199-6 |
Nicholson RV, Gillham RW and Reardon EJ. 1988. Pyrite oxidation in carbonate-buffered solution. Geochim. Cosmochim. Acta, 52: 1077-1085. DOI:10.1016/0016-7037(88)90262-1 |
Nordstrom DK and Southam G. 1997. Geomicrobiology of sulfide mineral oxidation. Reviews in Mineralogy, 35: 381-390. |
Osborne OD, Pring A and Lenehan CE. 2010. A simple colorimetric FIA method for the determination of pyrite oxidation rates. Talanta, 82(5): 1809-1813. DOI:10.1016/j.talanta.2010.07.086 |
Ouyang YT, Liu Y, Zhu RL, Ge F, Xu TY, Luo Z and Liang LB. 2015. Pyrite oxidation inhibition by organosilane coatings for acid mine drainage control. Minerals Engineering, 72: 57-64. DOI:10.1016/j.mineng.2014.12.020 |
Powell CJ, Jablonski A and Salvat F. 2005. NIST databases with electron elastic-scattering cross sections, inelastic mean free paths, and effective attenuation lengths. Surface and Interface Analysis, 37(11): 1068-1071. DOI:10.1002/(ISSN)1096-9918 |
Putnis A. 2014. Why mineral interfaces matter. Science, 343(6178): 1441-1442. DOI:10.1126/science.1250884 |
Qiu GZ, Xiao Q, Hu YH, Qin WQ and Wang DZ. 2004. Theoretical study of the surface energy and electronic structure of pyrite FeS2 (100) using a total-energy pseudopotential method, CASTEP. Journal of Colloid and Interface Science, 270(1): 127-132. DOI:10.1016/j.jcis.2003.08.028 |
Rickard D, Mussmann M and Steadman JA. 2017. Sedimentary sulfides. Elements, 13(2): 117-122. DOI:10.2113/gselements.13.2.117 |
Rimstidt JD and Vaughan DJ. 2003. Pyrite oxidation:A state-of-the-art assessment of the reaction mechanism. Geochimica et Cosmochimica Acta, 67(5): 873-880. DOI:10.1016/S0016-7037(02)01165-1 |
Rogowski AS and Pionke HB. 1984. Hydrology and Water Quality on Strip-mined Lands. U.S. Environmental Protection Agency
|
Rosso KM. 2001. Structure and reactivity of semiconducting mineral surfaces:Convergence of molecular modeling and experiment. Reviews in Mineralogy and Geochemistry, 42(1): 199-271. |
Sit PHL, Cohen MH and Selloni A. 2012. Interaction of oxygen and water with the (100) surface of pyrite:Mechanism of sulfur oxidation. The Journal of Physical Chemistry Letters, 3(17): 2409-2414. DOI:10.1021/jz300996c |
Somorjai GA. 1991. Chemistry in Two Dimensions: Surfaces. In: Zhen KJ, Yang H, Mao WQ, Wu ZY, Wang GJ, Cui XH, Guo CX, Wu TH, Bi YL, Li MX and Ji YS (Trans.). Changchun: Jilin University Press (in Chinese)
|
Stirling A, Bernasconi M and Parrinello M. 2003. Ab initio simulation of water interaction with the (100) surface of pyrite. The Journal of Chemical Physics, 118(19): 8917. DOI:10.1063/1.1566936 |
Sun HY, Chen M, Zou LC, Shu RB and Ruan RM. 2015. Study of the kinetics of pyrite oxidation under controlled redox potential. Hydrometallurgy, 155: 13-19. DOI:10.1016/j.hydromet.2015.04.003 |
Sun RS, Chan MKY and Ceder G. 2011. First-principles electronic structure and relative stability of pyrite and marcasite: Implications for photovoltaic performance. Physical Review B, 83(23): 235311 https://www.researchgate.net/publication/228475929_First-principles_electronic_structure_and_relative_stability_of_pyrite_and_marcasite_Implications_for_photovoltaic_performance
|
Sunagawa I. 1957. Variation in crystal habit of pyrite. Report of Geological Survey of Japan, 175: 1-47. |
Zhang YN, Hu J, Law M and Wu RQ. 2012. Effect of surface stoichiometry on the band gap of the pyrite FeS2(100) surface. Physical Review B, 85(8): 085314. DOI:10.1103/PhysRevB.85.085314 |
Zhao ZH, Zhang P, Chen YH, Yao Y, Wu YJ, Geng XH and Deng XZ. 2010. Study on oxidation of pyrite by in situ absorption spectroscopy. Spectroscopy and Spectral Analysis, 30(12): 3375-3378. |
Zhu JX, Xian HY, Lin XJ, Tang HM, Du RX, Yang YP, Zhu RL, Liang XL, Wei JM, Teng HH and He HP. 2018. Surface structure-dependent pyrite oxidation in relatively dry and moist air:Implications for the reaction mechanism and sulfur evolution. Geochimica et Cosmochimica Acta, 228: 259-274. DOI:10.1016/j.gca.2018.02.050 |
蔡元吉. 1990. 黄铁矿晶体形态模拟实验研究. 中国地质科学院南京地质矿产研究所所刊, 11(2): 33-45. |
陈光远, 孙岱生, 张立, 臧维生, 王健, 鲁安怀. 1987. 黄铁矿成因形态学. 现代地质, 1(1): 60-76. |
索莫杰GA. 1991.二维表面化学.见: 甄开吉, 杨洪, 茂魏诠, 吴志芸, 王国甲, 崔相浩, 郭纯孝, 吴通好, 毕颖丽, 李敏学, 稽玉书译.长春: 吉林大学出版社
|