 2018, Vol. 34
2018, Vol. 34


2. 中国科学院地球化学研究所, 环境地球化学国家重点实验室, 贵阳 550081;
3. 中国科学院大学, 北京 100049
2. State Key Laboratory of Environmental Geochemistry, Institute of Geochemistry, Chinese Academy of Sciences, Guiyang 550081, China;
3. University of Chinese Academy of Sciences, Beijing 100049, China
近年来,高分辨多接收杯电感耦合等离子体质谱仪(HR-MC-ICP-MS)的迅猛发展,极大推进和拓展了非传统稳定同位素地球化学的研究领域。同位素双稀释剂法(double spike technique, DS)、标准-样品交叉法(Standard-Sample Bracketing, SSB)和元素外标法(Element Doping, ED)构成了这一研究领域的三种主流分析技术(Yang, 2009)。同位素双稀释剂法作为一种内标校正方法,与Sr、Nd、Hf等同位素中设定如86Sr/88Sr=0.1194的固定值类似,可以准确校正样品纯化与仪器测试过程中的分馏。由于其测试同位素比值的高准确度、高精度和高度重现性,在非传统稳定同位素的研究领域中已得到日益广泛的关注和使用。同位素双稀释剂法最早由美国的Dodson (1963)提出,随后被陆续使用于Pb、Ca、Fe、Cr、Zn、Se、Mo、Cd、Hg、Ni、Sr等元素同位素组成的高精度分析(Compston and Oversby, 1969; Hamelin et al., 1985; Woodhead et al., 1997; Galer, 1999, Johnson and Beard, 1999; Johnson et al., 1999; Siebert et al., 2001; Gopalan et al., 2006; Ripperger and Rehkämper, 2007; Schoenberg et al., 2008; Zhu et al., 2008; Konter et al., 2008; Schmitt et al., 2009; Mead and Johnson, 2010; 李津等, 2011a, b; Gall et al., 2012; Millet et al., 2012; Neymark et al., 2014)。虽然本文DS法适于具有4个及4个以上元素的同位素组成分析(三同位素的双稀释剂法另文说明),但相对于SSB、ED法而言,依然具有较为突出的方法优势。首先DS法能够校正仪器测试同位素过程中的质量歧视效应(mass bias effect),使其分馏校正的准确度较高(Mead and Johnson, 2010);其次可用于检测地质或环境样品中低含量元素稳定同位素组成的微小变化(Woodhead et al., 1995),可获得自然样品中低含量元素的准确浓度值和高精度同位素比值(Mann et al., 2012);再次是DS法能够校正样品分离纯化过程中产生的同位素分馏,可简化分析纯化流程,降低待测元素的回收率要求,提高样品分析的成功率;最后是对于4个以上元素的同位素,双稀释剂法依然可用于稳定同位素非质量分馏研究和准确计算如ε40Ca过剩的问题,避免了元素外标法中因外标元素与待研究元素离子化效率或行为不同而引起的可能误差与误判。当然,在使用DS法的过程中,技术人员也会面临诸多问题。这些问题主要是待测元素同位素比值提取和双稀释剂剔除的算法问题;其次是双稀释剂间以及其与样品间的最佳混合比;最后还要考虑双稀释剂的纯度及其组成的准确标定等因素(Rudge et al., 2009)。
目前,对于两单稀释剂及双稀释剂与样品之间的最佳配比主要有两种途径可以获得:①是通过实测数据进行拟合;②是通过计算机数值模拟。早期使用双稀释剂法时,由于受到计算机发展的限制,多采用实测数据进行检验。Cumming (1973)利用铅同位素双稀释剂法对Pb同位素组成分析的实测数据表明,Pb同位素比值的误差(e)大小与两单稀释剂之间的配比(λ)和双稀释剂与样品混合配比(p)呈现一定的函数关系,这一认识为后期利用计算机进行数值模拟奠定了基础。Rudge et al. (2009)使用线性误差增量法和双稀释剂牛顿迭代算法对可用于DS法的几乎所有元素进行了数值模拟,并以57Fe-58Fe双稀释剂的实测数据为例,将实测和模拟的误差值投入e-p坐标系中均呈现出一致的U形曲线,由此确定了误差最小时所对应的λ、p值即为双稀释剂及其与样品间的最佳混合比。由于在57Fe-58Fe双稀释剂数值优化过程中λ、p值均为0.5,与实测数据拟合完全一致,验证了DS法数值模拟的可靠性。John (2012)将Monte-Carlo法与双稀释剂的几何迭代算法(Siebert et al., 2001)结合,也模拟出了最优的λ、P值,其结果与Rudge et al. (2009)所得出的结论基本一致。
国外有关同位素双稀释剂的算法经历了五十余年的发展,现已在国际上几个一流同位素实验室内广泛使用。相对而言,国内相关研究还刚刚起步。双稀释剂法应用的关键在于双稀释剂间的最优混合以及其与样品间的最佳配比。λ、P值的选择将会影响双稀释剂法校正结果的准确度和精密度(Rudge et al., 2009)。本文在Eugster et al. (1969)、Johnson et al.(1999)、Fantle and Bullen (2009)和Mann et al. (2012)等研究基础上,结合Johnson et al. (1999)关于双稀释剂的鸟巢迭代算法和Monte-Carlo法,对λ、P值进行了数值模拟,获得了最优的λ、P值,该值与Rudge et al. (2009)和John (2012)数值模拟的结果基本相同。在此基础上,未使用以往研究中给出的稀释剂与样品的浓度比值(p),而是接受Johnson教授的提议,使用稀释剂中某一同位素(如50Cr-54Cr双稀释剂中54Cr或50Cr)和样品中某一同位素(如53Cr或52Cr)的比值(54Cr稀释剂/52Cr自然样品)来模拟和厘定稀释剂与样品的最优比范围。本文主要以Cr和Mo同位素为例,数值模拟得到了Cr和Mo同位素双稀释剂组合的最优λ、P值以及它们所对应的误差大小,并以数值模拟为基础说明了双稀释剂的定量选择依据和稀释剂组成标定的流程,希望此文的报道能够使国内同行全面了解双稀释剂法的数学原理,为同位素双稀释剂技术的应用和推广起到抛砖引玉和借鉴的作用。
1 同位素双稀释剂技术的数学原理 1.1 双稀释剂技术的数学原理同位素双稀释剂法即向样品溶液中加入已知同位素组成的双稀释剂,经质谱测定得到样品与稀释剂混合液的测量值,然后利用同位素混合模型和指数分馏规律构建的双稀释剂公式,反复迭代剥离出稀释剂后即可得到样品同位素的真实比值。其数学原理是当两端元样品混合时,混合物中某同位素的量等于各端元中该同位素量与其所占比例的乘积之和(Rudge et al., 2009)。若双稀释剂与样品混合时,根据质量平衡原则,混合物中同位素的组成可表示为:

|
(1) |
其中p为双稀释剂在混合样品中的比例,q为自然样品在混合样品中所占的比例,分别如(2)和(3)所示。下标m、s和n分别标记为混合样品、稀释剂和自然样品。
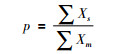
|
(2) |
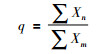
|
(3) |
(1) 和(2)式中iX为某元素中第i个同位素的丰度, ∑Xs、∑Xn和∑Xm为稀释剂、自然样品和混合样品的浓度。
如以相对简单的具有4个同位素的铬(Cr)元素为例,可简要说明双稀释剂相关公式的推导。假设待测Cr同位素的混合样品由N mol的自然样品和S mol的50Cr-54Cr双稀释剂混合而成,在自然样品和不纯的双稀释剂中50Cr、52Cr、53Cr、54Cr同位素的丰度分别为50Crn、52Crn、53Crn、54Crn和50Crs、52Crs、53Crs、54Crs。根据质量守恒原理,可由1式转化得到混合样品的同位素比值。若以(50Cr/52Cr)m为例,其表达式如4式,通过相关变换可得到(5)式,

|
(4) |
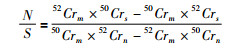
|
(5) |
分别从(5)式中等号右边的分子与分母中提出52Crs、52Crm、和52Crm、52Crn得到(6)式
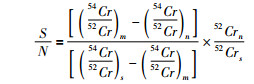
|
(6) |
同理,可得到N/S比与54Cr/52Cr、53Cr/52Cr比值的上述类似关系式。利用这些关系式,结合双稀释剂组成的初始值(54Cr/50Cr)s, trial公式(7)式,可以得到所选定双稀释剂如54Cr-50Cr的试验值公式(8)式, 该公式与Johnson et al.(1999)给出的硒同位素双稀释剂公式一致,其优点在于代码易编写,且考虑了双稀释剂成份的不纯性,并可通过混合样品的测量值与稀释剂同位素真值估算出仪器测量时的质量歧视效应。从(8)式观察,s和n的同位素比值均为固定值,因此,其质量歧视的大小受混合样品测量值的控制,可以有效反映质谱测量时的实时状态。
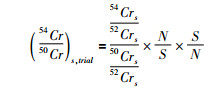
|
(7) |
由(6)式可知,(7)式中:

|
(8) |
经上述(6)式中N/S比可同理获得的53Cr/52Cr关系式,因此自然样品真实同位素比值的(53Cr/52Cr)n, trial提取公式如下式。

|
(9) |
利用上述公式(8)式和(9)式即可以得到同位素双稀释剂54/50Cr和自然样品53/52Cr的初试比值。如果以同位素标准替代上述公式中的自然样品,此时53/52Cr的初试比值即为同位素标准的真实比值,而双稀释剂54/50Cr比值也为其双稀释剂54Cr-50Cr的真实比值。
上述公式(8)和(9)可推而广之,对于任何具有4个或以上的同位素元素,如选取A1X, A2X作为双稀释剂(元素中丰度较低的同位素选作稀释剂),而将A1X,A2X,A3X,A4X用于同位素双稀释剂质量歧视效应的校正,可提取A3X/A4X的同位素比值,其中A3X丰度相对较高,同质异位素和多原子分子干扰较少。则公式(8)和(9)可转化为普遍性适用的双稀释剂通用公式(10)和(11)。其中公式(10)为双稀释剂初试比值提取公式,(11)为自然样品中的同位素初试比值的提取公式。
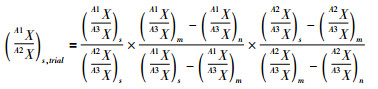
|
(10) |

|
(11) |
由于上述同位素比值在质谱仪的测定过程中,测得的同位素比值与真实值之间存在偏差,这一偏差即为质量歧视效应,且目前所使用的多接受杯等离子体质谱仪(MC-ICP-MS)对质量歧视效应的校正时普遍选择指数律(Albarède and Beard, 2004; Albarède et al., 2004)。Russell et al. (1978)和Hart and Zindler (1989)使用热电离质谱仪(TIMS)对Ca同位素进行研究时,发现指数律的拟合度最佳。指数规律的数学表达如下:
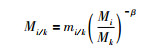
|
(12) |
其中mi/k为仪器对混合样品的测量值,以Cr为例,具体可表示为(53Cr/52Cr)m,Mi /k为样品的真实值,Mi为被测元素中第i个同位素的原子质量,Mk为参考同位素的原子质量,β为仪器的质量分馏因子。自然界中样品相对于同位素标准样品的分馏同样遵循指数分馏规律,则自然样品相对于标准的分馏律可表达为:
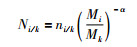
|
(13) |
其中Ni/k为样品测量值,ni/k为标准样品中第i个同位素与参考同位素的真实比值, α为自然样品相对于标准样品的分馏因子。这样,利用上述(10)、(11)、(12)和(13)给出的数学表达式,在双稀释剂试验值、仪器的分馏因子和自然样品的试验值进行交互迭代运算并至收敛,最终可得到自然样品的真实同位素组成。以Cr同位素双稀释剂法为例,即在50Cr-54Cr双稀释剂同位素质量歧视校正中,可由混合样品的测量值((50Cr/52Cr)m、(53Cr/52Cr)m、(54Cr/52Cr)m),双稀释剂的同位素比值((50Cr/52Cr)s、(53Cr/52Cr)s、(54Cr/52Cr)s、(54Cr/50Cr)s)、自然样品的同位素比值((50Cr/52Cr)n、(53Cr/52Cr)n、(54Cr/52Cr)n、(54Cr/50Cr)n)剥离出双稀释剂的试验值和自然样品的试验值,并求得相应的分馏因子。
对于稳定同位素而言,自然样品的同位素组成可由标准样品的同位素比值代替。但当待测元素含放射性成因的同位素如Pb、Sr等,则需对样品进行一次同位素组成测试,测量值可近似表示样品的同位素组成。双稀释剂计算的具体实施步骤为,首先通过双稀释剂比值的试验值与仪器分馏因子β交互迭代,不断地将混合样品测量值予以校正,直到两者收敛于某一固定值,如β=2.0等,此时双稀释剂比值也即将收敛至准确标定时的比值。其次将校正好的测量值带入(9)式或(11)式中,剥离出自然样品的同位素试验值,如(53Cr/52Cr)n, trial,并将其带入(13)式中,求出相对于原样品(标准样品)的同位素分馏值α,从而得到自然样品的所有同位素试验值。在实际工作中为节省运算时间,我们通常重复迭代至自然样品的同位素比值或α值收敛后才去计算我们需要提取的同位素比值。
需要注意的是,目前有关双稀释剂的算法种类有3~4种,如牛顿迭代、几何迭代或空间几何向量迭代等,大多采取解方程的类型,但数学原理基本一致,算法各有优缺点,但在优化范围内测试精度基本无差异。有关这些算法的优缺点比较将另文讨论。本文列出的算法中主要强调双稀释剂和仪器分馏因子的交互迭代与收敛,由于双稀释剂比值和仪器分馏因子β两个参数反复交互迭代的过程有如鸟类编织巢穴,故称之为鸟巢迭代。
1.2 双稀释剂技术的误差模型与估算在进行数值模拟双稀释剂中两单稀释剂间以及双稀释剂与样品间的最优配比时,需预设一定的误差模型(Rudge et al., 2009),此误差模型用于估算质谱测量时产生的误差大小。Dodson (1963)曾指出,质谱测定时的误差来源主要有三个方面:分别是电阻器噪声(Resistor Noise or Johnson Noise)、散粒效应(Shot Noise)和离子束强度的随意波动(Random Fluctuations in Ion Beam Intensity)。John (2012)利用双稀释剂法对海水同位素组成进行测定时,进行了实测误差值与理论误差值(完全由电阻器噪声和散粒效应模拟产生)的比较分析,得出δ57Fe,δ66Zn,δ114Cd的理论误差分别为实测误差的78%,85%,96%,进一步证实了电阻器噪声和粒射效应是仪器误差的主要来源。其中电阻器噪声(a)和散粒效应(d)为:
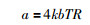
|
(14) |

|
(15) |
k为Boltzmann常数,k=1.3806504×10-23JK-1,b为频宽,即每秒可传送的离子数,R为电阻,预设为当前MC-ICP-MS标准配置前置放大器的1011Ω,T为质谱运行时的绝对温度,预设为300K,e为电子电荷,e=1.602176487×10-19C(库伦),b为传送周期t的倒数,默认时间t=8s。上述有些参数可依据所使用仪器条件适当修改。电阻噪声产生的绝对误差ρJohnson-noise与离子束强度无关,散粒效应产生的误差ρShot-noise与离子束强度的平方根呈正比关系。它们的表达式分别为:

|
(16) |
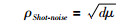
|
(17) |
其中,μ为某同位素在仪器中的离子强度,离子束强度的随意波动与同质异位素和同质量多原子离子的干扰强度有关。干扰较强时,波动范围较大。上述干扰所产生的误差表示为σisobar-interference。由此,对于某种同位素,质谱测定时所产生的误差可定义为:

|
(18) |
由于同质异位素和多原子离子干扰所产生的误差难以明确评估,其与仪器的状态(特别是灵敏度)和样品纯化的洁净度有关。对于理想的纯净溶液,众多产生干扰的元素可予以排除,此时仪器测试过程引入的误差接近电阻噪声和散粒效应所产生的误差。在模拟计算过程中可以不考虑同质异位素等干扰所引入的误差,σisobar-interference可预设为0。
模拟值(测量值)的误差求解是双稀释剂优化的关键。当误差最小时所对应的λ(某单稀释剂在双稀释剂中的比例)、p(双稀释剂在混合样品中的比例)值即为最优的配比值。目前,双稀释剂法的误差估算主要有两种方式,分别是线性误差增量法(Hamelin et al., 1985; Rudge et al., 2009; Yang, 2009)和Monte-Carlo法(Johnson and Beard, 1999; Mel’nikov, 2005; Fantle and Bullen, 2009,Lacan et al., 2010)。线性误差增量法涉及到众多的微分和矩阵运算,程序代码的编写较为复杂,但其具有运算快和数据易平滑的特点。相比较而言,Monte-Carlo法是适用于任一种算法的误差求解,代码易于编写,运算耗时与设置的数据点量密切相关,但都可预设在接受的范围内。本文使用了Monte-Carlo法对相关的误差进行求解。Monte-Carlo法的核心思想是输入一批均匀分布的数据,输出一组正态分布的数据。其具体步骤为,假定样品中某同位素的离子束强度为I,若令λ、P分别表示某单稀释剂在双稀释剂中的比例和双稀释剂在混合样品中的比例,以50Cr-54Cr双稀释剂为例,λ、P值的变化能够产生一系列的双稀释剂离子束IS和一系列混合样品离子束强度值IM:

|
(19) |
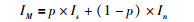
|
(20) |
其对应误差大小为σ,可根据误差模型计算,建立下列误差协方差矩阵。

|
(21) |
将离子束强度为I和误差协方差矩阵结合,经蒙特-卡洛法运算得到一组正态分布的信号值,同时求出各同位素的测量值,带入(10)~(13)的迭代公式中运算,查看其误差增量的大小。误差最小时所对应的λ、p值就是我们需要的最优配比值。
2 双稀释剂间及稀释剂与样品间配比的数值优化 2.1 双稀释剂间配比的数值优化利用1.2节中预设的误差模型与Monte-Carlo算法结合,可产生一系列的测量值,这些测量值经过(10)~(13)式的相关迭代运算,可以得到每次分馏校正的误差增量,通过对比误差增量值的大小来实现双稀释剂组成及与自然样品之间的配比优化。我们已知误差大小与双稀释剂间的配比(λ)和双稀释剂与样品混合时的配比(p)呈现一定的函数关系(Cumming, 1973)。若以50Cr-54Cr双稀释剂为例,λ可为50Cr或54Cr在50Cr-54Cr双稀释中所占的比例,p即为双稀释剂在双稀释剂与自然样品混合中所占的比例。当λ、p发生变化时,校正后得到的误差大小也随之变化,误差最小时所对应的λ、p即为双稀释剂间及其与自然样品的最优配比。在50Cr-54Cr同位素双稀释剂配比的优化中,改变λ、P值时,经Monte-Carlo算法模拟计算得到的误差曲线如图 1所示,(54Cr/50Cr)spike=0.67, 1, 1.5的误差曲线几乎重合,即(54Cr/50Cr)spike的值为0.67、1、1.5时,自然分馏因子α的误差几乎一致;而(54Cr/50Cr)spike=0.25或4时,α的误差值明显偏大。这说明(54Cr/50Cr)spike的值在0.67~1.5之间变化时,所产生的误差差异较小。换言之,也就是在配制双稀释剂时,最优的(54Cr/50Cr)spike值可以选择在0.67~1.5的比值范围内。在实际的双稀释剂操作过程中,为配制双稀释剂及其迭代程序计算的方便,我们通常取(54Cr/50Cr)spike的比值为1,这也就是为什么许多研究者使用的Cr同位素双稀释剂中两单稀释剂54Crspike/50Crspike比值在1左右波动的原因。同理,对于97Mo-100Mo双稀释剂,当(97Mo/100Mo)spike=0.5、1、1.5时模拟计算得到的误差曲线几乎一致,也就是自然分馏因子α的误差趋向一致;但当(97Mo/100Mo)spike=0.25、2或3时,α的误差值明显偏大。这说明在配制97Mo-100Mo双稀释剂时,最优的(97Mo/100Mo)spike混合比可以在0.5~1.5的范围内。实际操作过程中,为节省稀释剂和后续计算的方便,可以取(97Mo/100Mo)spike的比值为1。

|
图 1 基于Mento-Carlo误差模型的50Cr-54Cr双稀释剂在不同混合比下数值模拟优化结果图 Fig. 1 the numerical optimization for different mixing proportion of 50Cr-54Cr double spike based on Monte-Carlo error model |
不过这里需要强调的是,最优(54Cr/50Cr)spike值的范围可随(54Cr/50Cr)spike值选取的密集程度波动,并不是唯一的确定,但总体趋势不会有较大变化。研究者可以依据单稀释剂的成本和购买量,进行最优范围的最佳混合。此外,对于具有5个以上同位素的元素,双稀释剂间的配比也与提取的同位素比值对有关,如Cd元素具有8个稳定同位素,进行高精度分析时往往选择111Cd和113Cd作为双稀释剂,提取114/110Cd时其最佳比值为111Cd/113Cd≈1.25, 而当提取116/110Cd时其最佳比值为111Cd/113Cd≈1.50。此时应充分考虑仪器分馏因子与质量数间的关系,以及选择提取那些同位素比值对的必要性。我们在实际应用中,因考虑了Sn对116Cd、114Cd和112Cd同位素的干扰,以及116/110Cd具有较宽的质量范围,选取的双稀释剂111Cd/113Cd比值在1.50左右。
2.2 双稀释剂与自然样品间的配比优化双稀释剂与自然样品间的配比优化与双稀释剂间的配比优化类似,即为双稀释剂与自然样品以何种比例混合时,才能确保分馏校正过程中的误差最小。如图 1所示,当双稀释剂在混合样品中的比例p从0变化到1时,α所产生的误差大小是有所差异的,靠近0或1值时的误差较大,呈现U型的误差曲线,这种曲线使得研究者在寻找最小误差时所对应的p值更为容易。在实际应用中,由于优化的双稀释剂比通常在一定的范围内,且为了考虑后期迭代计算的方便和实验的易操作性,两单稀释剂的优化比一般取值为整数或带小数点的有理整数,如0.5、1等。图 2a是(54Cr/50Cr)spike=1时的误差曲线变化,该曲线是由三种不同计算方法求得的三组误差曲线,其中TZ-1,S-1分别由双稀释剂鸟巢迭代算法、几何迭代算法结合Monte-Carlo模拟而得,R-1为解方程组迭代算法结合线性误差增量法模拟而得,其中本次研究所采用方法计算得到的结果为TZ-1, 而S-1和R-1分别采用John (2012)和Rudge et al. (2009)所采用的方法。三种方法模拟得到最优p值的范围基本一致,最优范围均在0.15~0.55。使用Monte-Carlo法及线性误差增量法得到的α误差值存在一定差异,也就是Monte-Carlo法得到的α误差值明显小于线性误差增量法得到的α误差值,这与Rudge et al. (2009)的解释基本一致,也肯定了我们算法模拟的正确性。

|
图 2 50Cr-54Cr双稀释剂数值优化的模拟结果图 (a)显示了三种不同双稀释剂算法的比较,TZ-1为本文算法模拟结果,S-1为John (2012)、R-1为Rudge et al. (2009)的算法模拟结果; (b)是将横坐标中双稀释剂在混合样品中的比例更改为更为直观的表达方式,即为54Cr稀释剂/52Cr样品 Fig. 2 The diagram showing the results of numerical optimization for 50Cr-54Cr double spike (a) presents characteristics of our 50Cr-54Cr double spike optimization (TZ-1) and other optimization methods proposed by John (2102) (S-1) and Rudge et al. (2009) (R-1); (b) changed a more intuitive horizontal axis (54Crspike to 52Crsample)to instead of the proportion of double spike in spike-sample mix |
图 2a中可观察到双稀释剂与样品最优混合p值的范围。最佳p值的选取可以从0.15到0.55。在此范围内,α的误差相对较小。最佳p值的范围选取需考虑研究者所能容忍的误差大小,容忍的误差较大时p值的变化范围也可稍微扩大,此时也意味着研究者要求的方法分析精度稍微较低;反之,p值的变化范围可稍微缩小,分析精度也相对较高。理论计算时一般可给出最优的p值,即为曲线中α误差最小时所对应的p值,图 2a就可看出p在0.20~0.45时α误差相对最小,也就是双稀释剂在混合样品中的比例为20%~45%,自然样品在混合样品中的比例为55%~80%时所产生的误差不存在较大的变化。为避免稀释剂在使用过程中的过量加入,达到节省和准确加入的目的,我们建议使用双稀释剂中某一单稀释剂与样品中某一同位素的比值。如图 2b中,双稀释剂50Cr与54Cr的比值为1混合时,54Cr稀释剂/52Cr样品的比值范围在0.15~0.5间均可,引入的误差不存在较大差异,其它同位素的数值模拟也存在类似的曲线。对于97Mo-100Mo双稀释剂而言,模拟计算得到的p值为0.57,最优的取值范围是0.3~0.7。当使用100Mo稀释剂/98Mo样品进行实际样品操作时,在(97Mo/100Mo)spike为1的条件下,最优的100Mo稀释剂/98Mo样品是2.75,如在1.5~8间也是允许的范围,而有关此部分的数值模拟,因涉及专利申请的核心内容,我们在此不便作过多介绍。此外,需要说明的是,在确定双稀释剂最优配比的情况下,实际分析过程中应该确定双稀释剂与自然样品混合的最低(underspike)和最高(overspike)配比,明确混合的最佳有效范围,确保达到同位素比值的高精度分析。
3 同位素双稀释剂数值模拟的应用在同位素双稀释剂数值模拟的技术没有出现之前,同位素双稀释剂法的应用仅限于几个著名的同位素地球化学实验室。在这些实验室内,同位素双稀释剂的使用有着长期应用后积淀的经验性法则。但在进行双稀释剂间以及稀释剂与样品的混合配比时,仍然需要设计一系列的实验进行质谱的实际测试,寻找最佳的组合和配比。此过程中也必然浪费大量的稀释剂、人力和耗费机时,这也是一般地球化学实验室未能有效使用双稀释剂技术的主要原因。自Rudge et al. (2009)发表了同位素双稀释剂数值模拟的结果后,同位素双稀释剂技术开始逐渐推广。本文在此主要利用我们的同位素双稀释剂数值模拟结果,进行同位素双稀释剂选择及其混合后标定流程等重要前期步骤的详细说明,目的是有助于推动同位素双稀释剂技术在我国同位素地球化学界的广泛使用。
3.1 同位素双稀释剂的选择同位素双稀释剂的选择需要遵循一定的原则。这些原则可以从定性与定量两个方面说明。定性方面的选择(Johnson et al., 1999;Siebert et al., 2001):①是选为双稀释剂的两个同位素在自然样品中的丰度相对较低;②是选定的两个同位素在质谱测定过程中受同量异位素、多原子离子或基体效应的干扰相对较小或无干扰;③是选择作为稀释剂的同位素在实验室条件下易于纯化或分离,可以得到较为高纯的稀释剂,越为纯净的稀释剂在后期同位素测量的精度和绝对丰度计算方面也越有力。在定量方面,首先应该考虑的是双稀释剂在最优配比下校正所产生的误差相对较小(Galer, 1999; Rudge et al., 2009),且在λ、P最优值附近的较大区间内趋于平稳,不存在较大的波动。也就是说,在固定同位素双稀释剂比值的前提下,要保证样品与其中一稀释剂比值具有较大的范围而不致引起迭代运算中误差的较大波动。
以具有4个同位素的Cr元素和具有大于4个同位素的Mo元素为例,说明上述原则的如何使用。在自然界中,Cr有50Cr (4.34%)、52Cr (83.789%)、53Cr (9.501%)、54Cr (2.365%)这4种同位素,按照定性的原则可排除52Cr,其他双稀释剂的选择有50Cr-53Cr、50Cr-54Cr、53Cr-54Cr这3种可能。图 3显示了50Cr-53Cr、50Cr-54Cr、53Cr-54Cr双稀释剂在最优配比下校正的误差曲线图。50Cr-54Cr、53Cr-54Cr双稀释剂校正后所产生的误差最小,且误差值相当。50Cr-53Cr校正后的误差相对较大,且50Cr-53Cr双稀释剂在与自然样品混合时的最优配比选择范围较窄,并不利于实际的实验操作。因此,50Cr-53Cr双稀释剂并不是最优的选择,从定量角度上应该予以排除。50Cr-54Cr和53Cr-54Cr的误差曲线形态相似,但50Cr-54Cr双稀释剂的误差值变化相对平缓且范围较宽,在实验操作中可避免由于双稀释剂与样品配比的过高或过低造成的误差变化。因此,50Cr-54Cr双稀释剂用于同位素分馏校正时误差最小,且双稀释剂与自然样品混合时的配比范围较宽,可作为最优的双稀释剂选择。目前在众多Cr同位素双稀释剂的研究中(Schoenberg et al., 2008; Bonnand et al., 2011),50Cr-54Cr双稀释剂的使用均较为普遍。当然,根据研究需求,也可使用53Cr-54Cr双稀释剂,图 3也一样显示了其最佳的配比范围。

|
图 3 三组不同的Cr同位素双稀释剂数值优化结果比较 Fig. 3 the comparison of numerical optimization results for three different Cr isotopic double spike groups |
钼在自然界中有92Mo (14.836%)、94Mo (9.247%)、95Mo (15.920%)、96Mo (16.676%)、97Mo (9.555%)、98Mo (24.133%)、100Mo (9.633%)这7种稳定同位素。根据排列组合原理,可形成21(C72)对双稀释剂组合。若任意提取剩余5个同位素中的2个作为将要提取的比值,则最终可用于质量偏差校正的同位素组合有210种(C72×C52)。在实际应用中,需要考虑定性与定量方面的因素进行相关的取舍。国际上通用δ98/95Mo比值来表示样品中Mo同位素的组成,其原因在于95Mo、98Mo同位素的丰度适当,且基本上无干扰同位素的存在。因此,在双稀释剂校正过程中,当98Mo和95Mo两同位素被选定时,用于双稀释剂法校正的同位素组合降低到21种。根据上述定性原则的要求,92Mo、96Mo在样品中的丰度偏高,且易受92Zr+、96Zr+、96Ru+、60Ni36Ar+等同质异位数和多原子离子的干扰(Malinovsky et al., 2005),双稀释剂对的组合仅从94Mo、97Mo、100Mo中选取。如选定双稀释的组合为94Mo-97Mo、94Mo-100Mo和97Mo-100Mo,三组双稀释剂数值优化的结果如图 4所示。采用94Mo-100Mo双稀释剂法校正时,分馏因子α的误差最小。94Mo-97Mo、97Mo-100Mo所呈现的误差相近,但都略高于94Mo-100Mo。从优化的结果上观察,三组稀释剂组合均适用于仪器质量分馏的校正,并且94Mo-100Mo双稀释剂组合为最优组合。然而,对于94Mo-100Mo双稀释剂组合,未能推广使用的遗憾问题在于稀释剂中杂质元素Zr的存在。Zr同位素中94Zr的丰度(17.380%)仅次于90Zr (51.452%),在双稀释剂干扰校正中使用自然丰度90Zr作为基准值很难准确消除94Zr的干扰,这对94Mo稀释剂的准确标定存在潜在风险,地质样品也存在同样问题。因此,用于当前Mo同位素测定的双稀释剂多选择97Mo和100Mo。当然,如果存在研究的特殊需要或进行Mo同位素的非质量分馏研究,94Mo-100Mo双稀释剂依然是最佳选择,只不过此时稀释剂与样品都需要经过纯化分离,且地质样品中Zr、Fe(56Fe38Ar+或54Fe40Ar+)需要完全消除。

|
图 4 三组不同Mo同位素双稀释剂数值优化结果比较 Fig. 4 the comparison of numerical optimization results for three different Mo isotopic double spike groups |
元素的同位素稀释剂由于受到化学或物理分离提纯方法的限制,从供应商所购买的单稀释剂往往含有其它少量的同位素和杂质元素。在进行方法开发实验之前,首先要使用数值模拟结果了解两单稀释剂的混合比,其次均应对单和双稀释剂的同位素组成予以准确标定(Hamelin et al., 1985; Markey et al., 2003; Rudge et al., 2009)。对于购买的任何稀释剂而言,商家通常都提供稀释剂中的同位素丰度,但我们的实践证明,该值只能作为参考。单稀释剂的标定和要求相对简单,类似元素的浓度测定,双稀释剂的标定要求严格。单和双稀释剂组成的现行标定方法一般有三种:①是标准-样品交叉法标定,即在稀释剂测试前后分别对已知同位素组成的标准样品进行顺序测定,由指数律(12)式求出两次标准样品测试时的分馏因子β1和β2,将β1、β2的算术平均值作为稀释剂中各同位素比值的分馏因子,代入指数律(12)式对稀释剂的测量值予以校正,得到稀释剂的真实同位素组成;②是元素外标法标定,即往标定的稀释剂中加入与分析元素质量相近但互不干扰的元素进行标定。如在Mo的单或双稀释剂中加入Pd元素,假设Mo与Pd的仪器同位素分馏系数相同,即βMo=βPd,由(21)式可以得到稀释剂的同位素比值。该标定方法的缺陷是获得的双稀释剂比值完全依赖于假定的两元素同位素的分馏系数相同。如在标定Mo稀释剂组成的过程中,Pd同位素中104Pd/102Pd不是真实的同位素比值,或者βMo不能等同于βPd,需要使用经验性指数律公式对稀释剂组成和采用的同位素标准同时予以校正,此时获得的双稀释剂比值和同位素标样的比值都是假定的“真值”,但该值并不影响地质样品或其它标准样品相对于该同位素标准的δ比值,因为双稀释剂组成和同位素标样均使用Pd元素标定的,而δ只是表示相对比值的千分分馏。在实际的实验操作中,元素外标法标定稀释剂时应尽量选择已知同位素标准值且离子化效率或行为类似的元素。
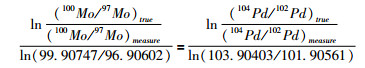
|
(22) |
在(22)式中,(104Pd/102Pd)true为Pd元素“假定”的同位素真实比值,(104Pd/102Pd)measure为对应同位素的测量值,(100Mo/97Mo)measure为稀释剂测量值,(100Mo/97Mo)true为稀释剂的假定“真实值”(李津等, 2011a, b)。③是同位素双稀释剂法标定,也就是往待标定的稀释剂中加入已知同位素比值的双稀释剂,经质谱测定,测量值由1.1节中的相关公式进行迭代校正,剥离出待标定稀释剂的真实比值。不过对没有双稀释剂使用经验的研究者而言,我们不建议选择该种方式来标定双稀释剂,较为简单的方法是选择①或②。在选择①或②方法标定双稀释剂时,直接有效的方式就是确定用于标定双稀释剂组成时标样中的同位素对,如标定97Mo-100Mo双稀释剂组成时,我们选择NIST SRM 3134中的100Mo/97Mo作为归一化的基准。
上述稀释剂的组成经初步标定完成后,还需进行一个重要步骤,那就是双稀释剂间以及双稀释剂与样品间的最优比混合。根据数值模拟优化、已有文献结果或经验性的认识,将标定好的单稀释剂按预先设定的双稀释剂最优比值混合后,按照上述方法标定,标定完毕后即将已标定好的双稀释剂与所采用的同位素标准按照稀释剂与样品的最优比混合,此时需要考虑干扰因素下的微调标定,并进行双稀释剂与不同样品浓度比(包括underspike和overspike)的最后检测,直到与双稀释剂已混合均匀的同位素标准中所有可能的(比值趋近0或收敛于0附近的某个固定值时,方可确定该双稀释剂的标定工作基本完成。同位素标定能否顺利的进行,必须依靠于准确地知道两单稀释剂和稀释剂与样品之间的配比值。在开发一些新的同位素双稀释剂方法时,由于缺乏前人研究的参考,或者稀释剂没有准确的标定之前,也很难通过实验获取稀释剂之间和稀释剂与样品之间的最优配比范围。这时稀释剂成分的不确定性,会增加误差的引入量,实测时难以区分是否由不同稀释剂比或者稀释剂成分不准而带入的误差。此时,双稀释剂的数值优化便显的极为重要,优化配比值的提供能有效地缩短双稀释剂标定的时间和节省大量的稀释剂。
4 结论随着多接收器电感耦合等离子质谱仪(MC-ICP-MS)在地球科学、环境科学及生命科学中的使用和普及,双稀释剂法的优势将会在非传统稳定同位素的研究领域中愈发明显。虽然双稀释剂法有其内在的固有缺点,但其能够同时校正样品分离纯化和仪器质量分馏的特征,使其具有简化化学分离流程和显著提高样品分析效率的明显优势。本文在诸多国外学者使用同位素双稀释剂法的基础上,以Cr和Mo同位素为例,对双稀释剂法的数学原理作了全面系统的介绍,详细说明了双稀释剂中单稀释剂间及双稀释剂与自然样品混合等最佳配比的数值优化过程中,给出了案例中50Cr-54Cr双稀释剂最优的λ、P值范围,为下一步的系列实验验证起到了指导作用。在双稀释剂数值模拟的基础上,讨论了同位素双稀释剂的选择原则,力图让研究者了解如何进行双稀释剂的最佳选择,并给出了双稀释剂选择的具体原则与方法。同时,就双稀释剂如何进行准确的标定和进行后期仪器上的实践验证,也给出了具体的建议。希望此文能够对国内同行了解、使用和推广同位素双稀释剂技术起到抛砖引玉和借鉴的作用。
致谢 本文工作的完成和发表得益于美国伊利诺伊大学香槟分校地质系Johnson教授、中国科学院地质与地球物理所吴福元研究员的鼓励和讨论,也得到中国地质科学院地质研究所朱祥坤研究员、中国科学院广州地球化学研究所张兆峰研究员、德国马普研究所Galer博士、英国剑桥大学Rudge博士以及两位匿名审稿人的建议和帮助,谨此致谢。
Albarède F and Beard B. 2004. Analytical methods for non-traditional isotopes. Reviews in Mineralogy and Geochemistry, 55(1): 113-152. DOI:10.2138/gsrmg.55.1.113 |
Albarède F, Telouk P, Blichert-Toft T, Boyet M, Agranier A and Nelson B. 2004. Precise and accurate isotopic measurements using multiple-collector ICPMS. Geochimica et Cosmochimica Acta, 68(12): 2725-2744. DOI:10.1016/j.gca.2003.11.024 |
Bonnand P, Parkinson IJ, James RH, Karjalainen AM and Fehr MA. 2011. Accurate and precise determination of stable Cr isotope compositions in carbonates by double spike MC-ICP-MS. Journal of Analytical Atomic Spectrometry, 26(3): 528-535. DOI:10.1039/c0ja00167h |
Compston W and Oversby VM. 1969. Lead isotopic analysis using a double spike. Journal of Geophysical Research, 74(17): 4338-4348. DOI:10.1029/JB074i017p04338 |
Cumming GL. 1973. Propagation of experimental errors in lead isotope ratio measurements using the double spike method. Chemical Geology, 11(3): 157-165. DOI:10.1016/0009-2541(73)90014-4 |
Dodson MH. 1963. A theoretical study of the use of internal standards for precise isotopic analysis by the surface ionization technique Part Ⅱ:Error relationships. Journal of Physics E:Scientific Instruments, 40(6): 289-295. DOI:10.1088/0950-7671/40/6/307 |
Eugster O, Tera F and Wasserburg GJ. 1969. Isotopic analyses of barium in meteorites and in terrestrial samples. Journal of Geophysical Research, 74(15): 3897-3908. DOI:10.1029/JB074i015p03897 |
Fantle MS and Bullen TD. 2009. Essentials of iron, chromium, and calcium isotope analysis of natural materials by thermal ionization mass spectrometry. Chemical Geology, 258(1-2): 50-64. DOI:10.1016/j.chemgeo.2008.06.018 |
Galer SJG. 1999. Optimal double and triple spiking for high precision lead isotopic measurement. Chemical Geology, 157(3-4): 255-274. DOI:10.1016/S0009-2541(98)00203-4 |
Gall L, Williams H, Siebert C and Halliday A. 2012. Determination of mass-dependent variations in nickel isotope compositions using double spiking and MC-ICPMS. Journal of Analytical Atomic Spectrometry, 27(1): 137-145. DOI:10.1039/C1JA10209E |
Gopalan K, Macdougall D and Macisaac C. 2006. Evaluation of a 42Ca-43Ca double-spike for high precision Ca isotope analysis. International Journal of Mass Spectrometry, 248(1-2): 9-16. DOI:10.1016/j.ijms.2005.09.009 |
Hamelin B, Manhes G, Albarede F and Allègre CJ. 1985. Precise lead isotope measurements by the double spike technique:A reconsideration. Geochimica et Cosmochimica Acta, 49(1): 173-182. DOI:10.1016/0016-7037(85)90202-9 |
Hart SR and Zindler A. 1989. Isotope fractionation laws:A test using calcium. International Journal of Mass Spectrometry and Ion Processes, 89(2-3): 287-301. DOI:10.1016/0168-1176(89)83065-4 |
John SG. 2012. Optimizing sample and spike concentrations for isotopic analysis by double-spike ICPMS. Journal of Analytical Atomic Spectrometry, 27(12): 2123-2131. DOI:10.1039/c2ja30215b |
Johnson CM and Beard BL. 1999. Correction of instrumentally produced mass fractionation during isotopic analysis of Fe by thermal ionization mass spectrometry. International Journal of Mass Spectrometry, 193(1): 87-99. DOI:10.1016/S1387-3806(99)00158-X |
Johnson TM, Herbel MJ, Bullen TD and Zawislanski PT. 1999. Selenium isotope ratios as indicators of selenium sources and oxyanion reduction. Geochimica et Cosmochimica Acta, 63(18): 2775-2783. DOI:10.1016/S0016-7037(99)00279-3 |
Konter JG, Pietruszka AJ and Hanan BB. 2008. Development of a58Fe-57Fe double spike for Fe isotopic analysis using a Nu Plasma 1700 MC-ICP-MS. Geochimica et Cosmochimica Acta, 72(12): A489. |
Lacan F, Radic A, Labatut M, Jeandel C, Poitrasson F, Sarthou G, Pradoux C, Chmeleff J and Freydier R. 2012. High-precision precision determination of the isotopic composition of dissolved iron in iron depleted seawater by double spike multicollector--ICPMS. Analytical chemistry, 82(17): 7103-7111. |
Li J, Zhu XK and Tang SH. 2011a. Mo isotope measurements using double spike techniques. Acta Geoscientica Sinica, 32(5): 601-609. |
Li J, Zhu XK and Tang SH. 2011b. The application of double spike in non-traditional stable isotopes:A case study on Mo isotopes. Rock and Mineral Analysis, 30(2): 138-143. |
Malinovsky D, Rodushkin I, Baxter DC, Ingri J and Öhlander B. 2005. Molybdenum isotope ratio measurements on geological samples by MC-ICPMS. International Journal of Mass Spectrometry, 245(1-3): 94-107. DOI:10.1016/j.ijms.2005.07.007 |
Mann JL, Vocke Jr RD and Kelly WR. 2012. Determination of low-level (sub-microgram) sulfur concentrations by isotope dilution multi-collector inductively couple plasma mass spectrometry using a 33S spike and internal normalization for mass bias correction. Rapid Communications in Mass Spectrometry, 26(10): 1175-1180. DOI:10.1002/rcm.6214 |
Markey R, Hannah JL, Morgan JW and Stein HJ. 2003. A double spike for osmium analysis of highly radiogenic samples. Chemical Geology, 200(3-4): 395-406. DOI:10.1016/S0009-2541(03)00197-9 |
Mead C and Johnson TM. 2010. Hg stable isotope analysis by the double-spike method. Analytical and Bioanalytical Chemistry, 397(4): 1529-1538. DOI:10.1007/s00216-010-3701-0 |
Mel'nikov NN. 2005. Errors of the double spiking technique in the isotopic analysis of common lead. Geochemistry International, 43(12): 1228-1234. |
Millet MA, Baker JA and Payne CE. 2012. Ultra-precise stable Fe isotope measurements by high resolution multiple-collector inductively coupled plasma mass spectrometry with a 57Fe-58Fe double spike. Chemical Geology, 304-305: 18-25. DOI:10.1016/j.chemgeo.2012.01.021 |
Neymark LA, Premo WR, Mel'nikov NN and Emsbo P. 2014. Precise determination of δ88Sr in rocks, minerals, and waters by double-spike TIMS:A powerful tool in the study of geological, hydrological and biological processes. Journal of Analytical Atomic Spectrometry, 29(1): 65-75. DOI:10.1039/C3JA50310K |
Ripperger S and Rehkämper M. 2007. Precise determination of cadmium isotope fractionation in seawater by double spike MC-ICPMS. Geochimica et Cosmochimica Acta, 71(3): 631-642. DOI:10.1016/j.gca.2006.10.005 |
Rudge JF, Reynolds BC and Bourdon B. 2009. The double spike toolbox. Chemical Geology, 265(3-4): 420-431. DOI:10.1016/j.chemgeo.2009.05.010 |
Russell WA, Papanastassiou DA and Tombrello TA. 1978. Ca isotope fractionation on the Earth and other solar system materials. Geochimica et Cosmochimica Acta, 42(8): 1075-1090. DOI:10.1016/0016-7037(78)90105-9 |
Schmitt AD, Galer SJG and Abouchami W. 2009. High-precision cadmium stable isotope measurements by double spike thermal ionisation mass spectrometry. Journal of Analytical Atomic Spectrometry, 24(8): 1079-1088. DOI:10.1039/b821576f |
Schoenberg R, Zink S, Staubwasser M and Von Blanckenburg F. 2008. The stable Cr isotope inventory of solid Earth reservoirs determined by double spike MC-ICP-MS. Chemical Geology, 249(3-4): 294-306. DOI:10.1016/j.chemgeo.2008.01.009 |
Siebert C, Nägler TF and Kramers JD. 2001. Determination of molybdenum isotope fractionation by double-spike multicollector inductively coupled plasma mass spectrometry. Geochemistry, Geophysics, Geosystems, 2(7): 2000G. |
Woodhead JD, Volker F and McCulloch MT. 1995. Routine lead isotope determinations using a lead-207-lead-204 double spike:A long-term assessment of analytical precision and accuracy. Analyst, 120(1): 35-39. DOI:10.1039/AN9952000035 |
Woodhead JD and Hergt JM. 1997. Application of the 'double spike' technique to Pb-isotope geochronology. Chemical Geology, 138(3-4): 311-321. DOI:10.1016/S0009-2541(97)00013-2 |
Yang L. 2009. Accurate and precise determination of isotopic ratios by MC-ICP-MS:A review. Mass Spectrometry Reviews, 28(6): 990-1011. DOI:10.1002/mas.v28:6 |
Zhu JM, Johnson TM, Clark SK and Zhu XK. 2008. High precision measurement of selenium isotopic composition by hydride generation multiple collector inductively coupled plasma mass spectrometry with a 74Se-77Se double spike. Chinese Journal of Analytical Chemistry, 36(10): 1385-1390. DOI:10.1016/S1872-2040(08)60075-4 |
李津, 朱祥坤, 唐索寒. 2011a. 钼同位素比值的双稀释剂测定方法研究. 地球学报, 32(5): 601-609. |
李津, 朱祥坤, 唐索寒. 2011b. 双稀释剂法在非传统稳定同位素测定中的应用-以钼同位素为例. 岩矿测试, 30(2): 138-143. |