
A survey of deep learning-based drug-target interaction prediction
-
摘要: 新药物研发是一项耗时、耗力、耗资的复杂工程,整体成功率低于10%。药物−靶标相互作用预测是药物筛选和药物重定位的关键环节。准确的药物−靶标相互作用预测可有效缩小候选药物分子筛选范围,加速药物研发进程。传统实验方法研究药物−靶标相互作用耗时长、成本高且伴有一定的盲目性,难以进行大规模的药物−靶标相互作用识别工作。近年来,将机器学习尤其是深度学习技术用于药物−靶标相互作用预测成为主流研究。尽管在过去10年有大量的研究工作纷纷涌现,药物−靶标相互作用预测仍然是物质密集型和长期性的工作,对研究者来说仍具有挑战性。本文梳理近年来基于深度学习的药物−靶标相互作用预测研究工作,归纳总结现有工作的研究方法、评价指标和使用的数据资源,分析现有工作的不足并提出展望。本文的研究目的是帮助药物研发领域研究者全面了解深度学习在药物−靶标相互作用预测领域的最新研究进展,从而提高研究效率和研究质量。Abstract: The development of novel drugs is a time-consuming, labor-consuming, and costly process with the overall success rate no more than 10%. The prediction of drug-target interactions (DTIs) is fundamental for drug screening and drug repositioning. Accurate DTI prediction can significantly narrow down the screening of drug candidates and accelerate the drug discovery process. The traditional experimental method for identifying DTIs is tedious and expensive and accompanied by certain blindness, which restricts it from large-scale DTI identification. Recently, applying machine learning especially deep learning techniques to DTI prediction has become the mainstream. Although a series of methods have been proposed in the last decade, DTI prediction is still a material-intensive and long-term work, and is challenging to researchers. In this survey, we review literature related to DTI prediction, and summarize the methodologies, evaluation indicators, and data sources used in these works. We also analyze the shortcomings of existing works and propose future prospects. Our motivation is to help researches dedicated to drug discovery and development to have a comprehensive understanding on the latest progress of DTI prediction so as to improve their research efficiency and research quality.
-
药物研发历史悠久,在史前时期便已存在[1]。时至今日,药物研发仍是一项不断实验,反复试错的过程。在20世纪初,新药的发现仍属于偶然事件。Paul Ehrlich为治疗梅毒素建立的新疗法开创了现代药物发现的先河,人们开始使用系统的筛选方法研究新药[1]。新药研发是一项耗时耗资耗力的复杂工程,起始于某种人们感兴趣的疾病,由市场规模和临床需求决定。新药研发包括疾病相关基因的发现、靶标识别、靶标确证、先导化合物发现、先导化合物优化、临床前研究以及临床试验等关键步骤,直到证明分子足够安全有效才能获批上市,历时10~17年,花费20亿~30亿美金[2-5],但仅有10%的药物能通过第一阶段临床试验,其他的药物因具有较高的毒副作用或较低的药效而被淘汰[4]。对肿瘤类复杂疾病,成功率更低,仅有5%的药物通过第一阶段临床试验[6]。制药行业的反摩尔定律使得新药的研发成本不断提高,成功率不断下降,研发效率也变得越来越低[3]。从全球来看,自1950年以来,每10亿美元研发投入获得批准的新药数量几乎每9年减少一半。2019年,英国德勤会计师事务所的一份报告指出,当年新药研发的投资回报率仅为1.8%,而10年前回报率则高达10%左右[7]。从1993年至今,FDA(United States food and drug administration)每年批准的新药数量增长十分缓慢[8]。传统药物研发模式高度依赖药物研发人员的个人经验与创造力,周期长、成本高、效率低、风险大。因此,迫切需要新技术、新模式来加快药物研发的进程。
进入21世纪后,整个人类的基因组及许多其他物种的基因组已被阐明,生命科学、计算机科学和人工智能(artificial intelligence,AI)快速发展。第二代高通量测序技术、冷冻电镜等为代表的各类组学技术飞速发展,使得生命科学研究获得了强大的数据产出能力,包括基因组学(基因结构和功能的分析)、蛋白质组学(研究蛋白质结构和功能)、代谢组学(细胞过程的代谢“指纹”)等生物学数据,为人工智能在新药研发领域发挥价值提供了丰富的原材料。自2006年以来,在大计算能力和深度学习(deep learning,DL)的推动下,人工智能发展迅速,进入第三次浪潮,开始广泛应用于计算机视觉[9-10]、自然语言处理[11]、推荐系统[12-13]、医疗图像分析[14]等领域,也促使很多创业公司、互联网科技企业以及科研院所开始探索人工智能在药物研发领域中的应用。与此同时,全球AI制药投融资呈指数增长,国家和地方政府也纷纷出台一系列政策鼓励和引导企业将人工智能技术应用于药物研发。行业需求下,大数据、人工智能、计算技术的快速发展以及资本大力助推等因素的共同作用下,全球AI/计算制药领域掀起发展热潮。根据BBC数据显示,人工智能技术在医疗健康产业所有的应用中,药物研发在市场规模和增长速度两个方面都拔得头筹。国际上,几乎每一家药企都至少与一家人工智能企业开展了紧密合作。例如,阿斯利康公司仅在2019年就发布了65篇人工智能相关的药物研发文献[15]。2020年,Google DeepMind的AlphaFold II[16]发布取得重大突破,解决了困扰科学家50年的生物学难题——蛋白质折叠。
人工智能技术以数据为基础,既能够直接赋能药物研发,也可以通过赋能计算间接驱动药物研发,可以在很大程度上实现人类在体能、智力等全方位增强,同时也可以避免人工偏向性,减少人力成本。人工智能具有技术延展性优势,随着数据量增加、质量提升以及算法的训练突破,可以实现叠加式进步发展。其目标不局限于实现降本增效,尽量减少合成化合物进行实验的次数,更大的目标在于试图通过新技术实现平台化、规模化产出新药的能力[7]。据北京大学前沿交叉学科研究院定量生物学中心裴剑锋教授介绍,融合运用人工智能技术,可为前期新药研发阶段节约40%~50%的时间,每年节省约260亿美元的化合物筛选成本和280亿美元的临床实验费用,人工智能正成为药物研发的重要新工具。目前人工智能技术在药物研发领域还处于“单点突破”阶段,广泛应用于药物研发的多个环节,涉猎靶点发现、分子生成、活性预测、ADMET(absorption, distribution, metabolism, excretion,and toxicity)性质预测、化合物合成、虚拟药物筛选等。其中,分子生成、分子活性预测以及ADMET性质预测是药物发现的核心环节,关注较多。
本文重点研究人工智能尤其是深度学习技术在药物−靶标相互作用(drug-target interaction,DTI)预测方面的研究进展。理解药物−靶标相互作用是生物化学、生物物理和分子生物学的核心问题[17]。药物−靶标相互作用预测是药物筛选和药物重定位的关键环节,可有效缩小候选药物分子的搜索范围。亲和力(affinity)衡量药物和靶标之间的相互作用强度,较高的结合亲和力使药物能够在低剂量情况下产生预期疗效,降低毒副作用风险,增加实用性[18]。人类蛋白质组大约包含70 000个蛋白质序列,可合成的化学分子有1060个。传统实验方法研究药物−靶标相互作用耗时长、成本高且伴有一定的盲目性,难以进行大规模的药物−靶标相互作用识别工作。将人工智能技术尤其是深度学习技术运用到药物−靶标相互作用的挖掘、预测,正成为人工智能领域和计算生物学领域的一个研究热点,在学术界和工业界得到广泛关注。尽管过去几年大量的研究工作纷纷涌现,药物−靶标相互作用预测仍然是物质密集型和长期性的工作,对研究者来说仍具有挑战性。本文梳理近年来基于机器学习/深度学的药物−靶标相互作用预测研究工作,归纳总结现有工作的研究方法、评价指标和使用的数据资源,分析现有工作的不足并提出展望。本文的研究目的是帮助药物研发领域研究者全面了解深度学习在药物−靶标相互作用预测领域的最新研究进展,从而提高研究效率和研究质量。
1. 药物−靶标相互作用预测研究意义
识别潜在的药物和靶标是药物研发初始阶段的关键步骤,可以大幅度缩减药物研发周期和研发成本。药物−靶标相互作用在许多重要生物过程中起到决定性作用,如酶催化反应、信号通路、细胞信号传导、转录、代谢和免疫。药物又称配体,通常是小分子化合物或生物制剂,用于控制、预防、治疗和诊断疾病。靶标是指人体内与某些疾病相关的具有特定化学性质且能够与药物进行特异性结合的生物分子,如蛋白质和核酸。大多数的药物靶标是蛋白质。蛋白质是器官和组织的重要组成部分,参与所有的重要生物过程,如输送氧气、催化反应、传送生物信号等。绝大多数药物靶标蛋白属于4种大分子,即G蛋白偶联受体(G-protein-coupled receptor,GPCR)(以此为靶点的药物约占44%)、酶(enzyme)(以此为靶点的药物约占29%)、载体蛋白(又称转运体(transporter))(以此为靶点的药物约占15%)和离子通道(ion channel)。
如图1所示,药物和靶标的相互作用关系类似于钥匙和锁,相互作用发生在靶标蛋白的特定位置,称为结合位点(binding site)或者结合口袋(binding pocket),是蛋白质表面的凹陷。结合位点通常出现在蛋白质表面大且深的口袋中,但也会出现在一些浅层的裂缝中。如图2所示,药物−靶标相互作用的过程就是药物分子与其具有相同结构和功能特性的活性生物分子位点结合,生成新的产物,人体吸收该产物,从而达到治疗疾病的目的[19]。实际上,如果药物与结合位点具有较高的结合亲和力并产生有效的治疗作用,该位点就是可药性位点[20]。亲和力衡量药物和靶标之间的相互作用强度,用抑制常数(inhibition constant,
$ {K}_{i} $ )、解离常数(dissociation constant,$ {K}_{d} $ )、半数抑制浓度(half-maximal inhibitory concentration,$ {\mathrm{I}\mathrm{C}}_{50} $ )、半数有效浓度(half-maximal inhibitory effective concentration,$ {\mathrm{E}\mathrm{C}}_{50} $ )等衡量[21]。药效学(pharmacodynamics)研究发现,当药物进入人体内与靶标发生相互作用时,亲和力直接反映了药物的临床疗效[15]。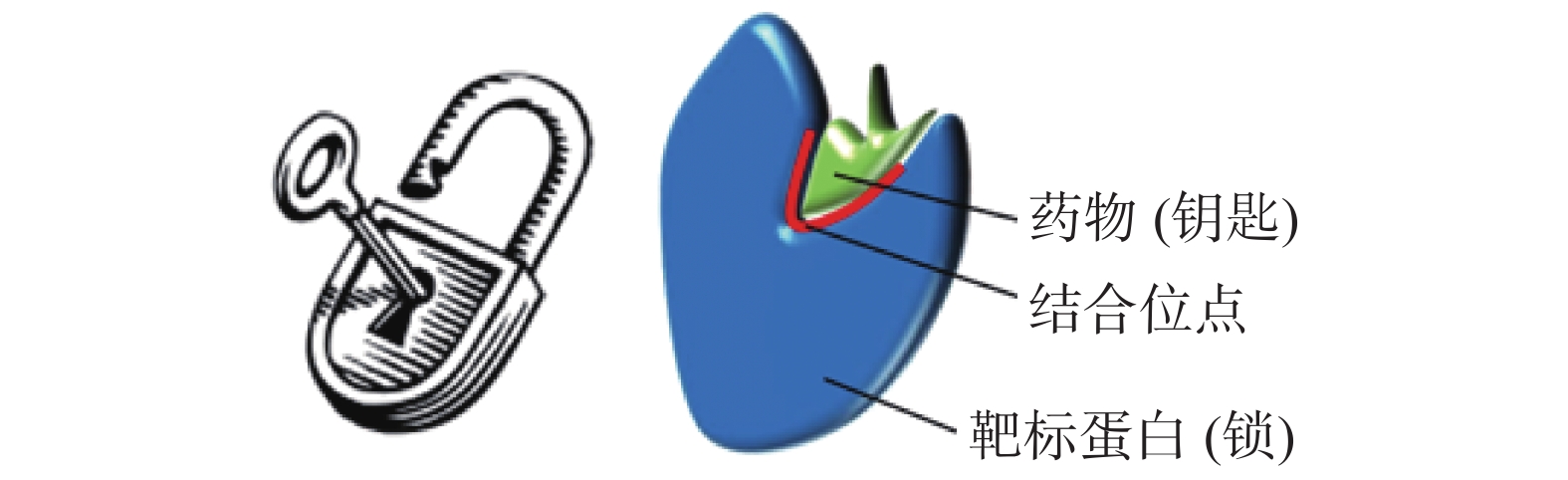 图 1 药物−靶标相互作用类比示意Fig. 1 Illustration of drug-target interaction
图 1 药物−靶标相互作用类比示意Fig. 1 Illustration of drug-target interaction 下载:
全尺寸图片
下载:
全尺寸图片
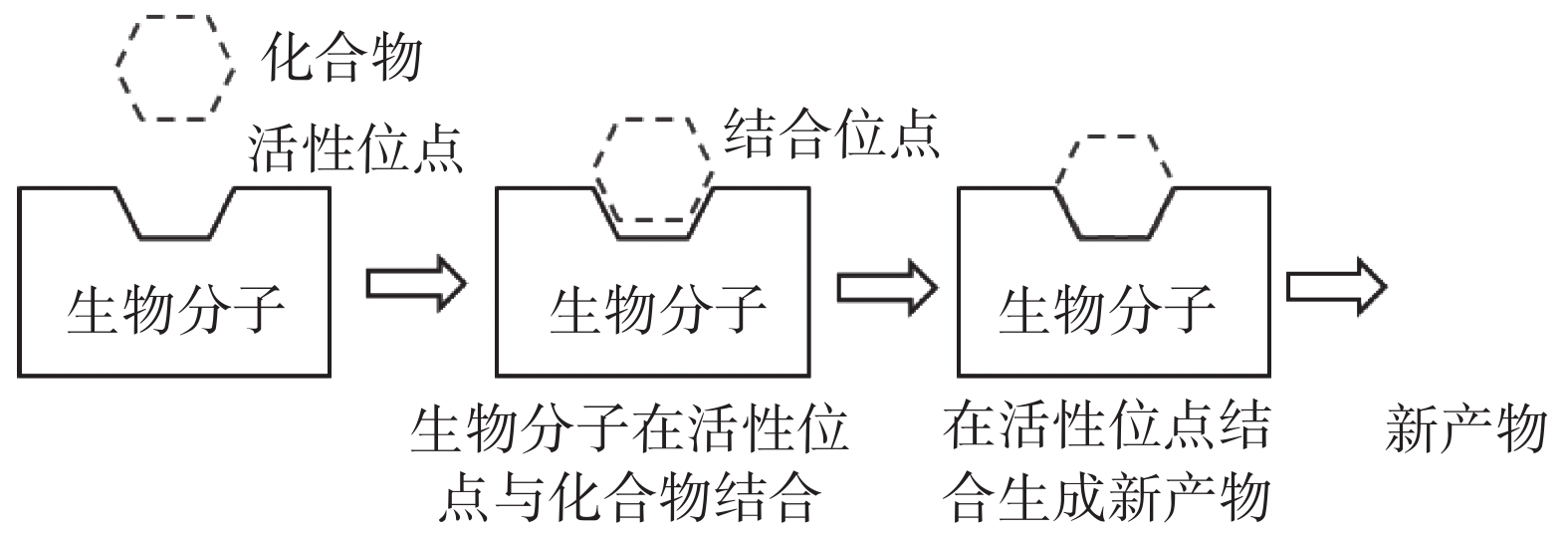 图 2 药物−靶标相互作用过程Fig. 2 Process of drug-target interaction下载:
全尺寸图片
图 2 药物−靶标相互作用过程Fig. 2 Process of drug-target interaction下载:
全尺寸图片
药物−靶标相互作用预测也是药物重定位(drug repositioning)的研究基础[3-4,22-28]。药物重定位是从已知药物中找到可以治疗其他疾病的药物,这些药物包括已经批准并用于日常临床环境中的药物,也包括由于临床试验失败或项目终止而停止继续研究的药物,在2004年由Ashburn和Thor首次提出[2]。计算机辅助药物重定位的快速发展和广泛使用得益于两方面因素[4]。第一,大量的高通量数据可以获得,包括基因组学、蛋白组学、化学蛋白组学和表型组学。因此,除了表征疾病的显型(phenotype)数据和药物信息,整个路径图(pathway map)都可以得到。第二,计算机和数据科学的发展,使研究者可以研发高效快捷的药物重定位算法,进行实验数据分析和结果保存。
可进行重定位的药物高达75%[29],在美国,超过30%的新批准药物都是重定位药物[30-31]。西地那非(Sildenafil)(伟哥(Viagra))、反应停(Thalidomide)、雷洛昔芬(Raloxifene)(易维特(Evista))等是药物重定位成功的典型案例[4,32]。药物重定位的目的主要包括两个方面,第一,扩展药物的应用范围或延长药物专利生命周期,增加利润收入。据估计,药物重定位平均成本为3亿美元[29]。因为已知药物已经包含临床前研究信息和临床信息(药代动力学、药效学和毒性),因而研发风险低,可以快速进入后临床试验阶段,大大降低研发周期和研发成本,可以在短期内获得投资回报。第二,治疗罕见疾病或突发疾病[33]。《FDA罕见用药法案》定义的罕见疾病(低于20万美国患病率的疾病)超过7 000种,95%以上的疾病没有相应的治疗药物[32-33]。如果对罕见疾病进行新药研发,可供研究样本有限,且研发成本不可估量,药物重定位可有效缓解这一困境。
药物可以参与多种生物过程并发挥多种生物功能,这为药物重定位提供依据[3,19]。第一,一个药物会与多个蛋白质发生相互作用,又称脱靶(off-target)反应或多药理学(polypharmacology),并产生一系列出人意料的反应。如疱疹药物BVDU会与病毒性胸腺嘧啶激酶(viral thymidine kinase)和与癌症密切相关的热休克蛋白(heat shock protein)Hsp27发生相互作用。识别出已知药物的脱靶蛋白就有机会为该药物重新定位(图3(b))。第二,靶标自身会在多重生物过程中发挥作用,如伟哥的重定位来自酶PDE5的第二功能——参与勃起过程(图3(c))。第三,药物起初只针对某种适应症(疾病)进行优化和临床试验,并未对所有可能的适应症(疾病)进行实验,这就意味着该药物在临床实践中可能会对其他疾病产生治疗效果(图3(d))。例如两种分子结构不同的抗偏头疼药物利坦色林(Ritanserin)和麦角胺(Ergotomine)都与靶标5-HT2C相结合。又如抗癌药物伊布替尼(Ibrutinib)与其靶标CDPK1的结合位置与药物帕唑帕尼(Pazopanib)和VEGFR2的结合位置相似,伊布替尼(Ibrutinib)与其靶标 CDPK1 的结合位置与药物帕唑帕尼(Pazopanib)和 VEGFR2 的结合位置相似,伊布替尼也会抑制 VEGFR2 达到抗癌效果。
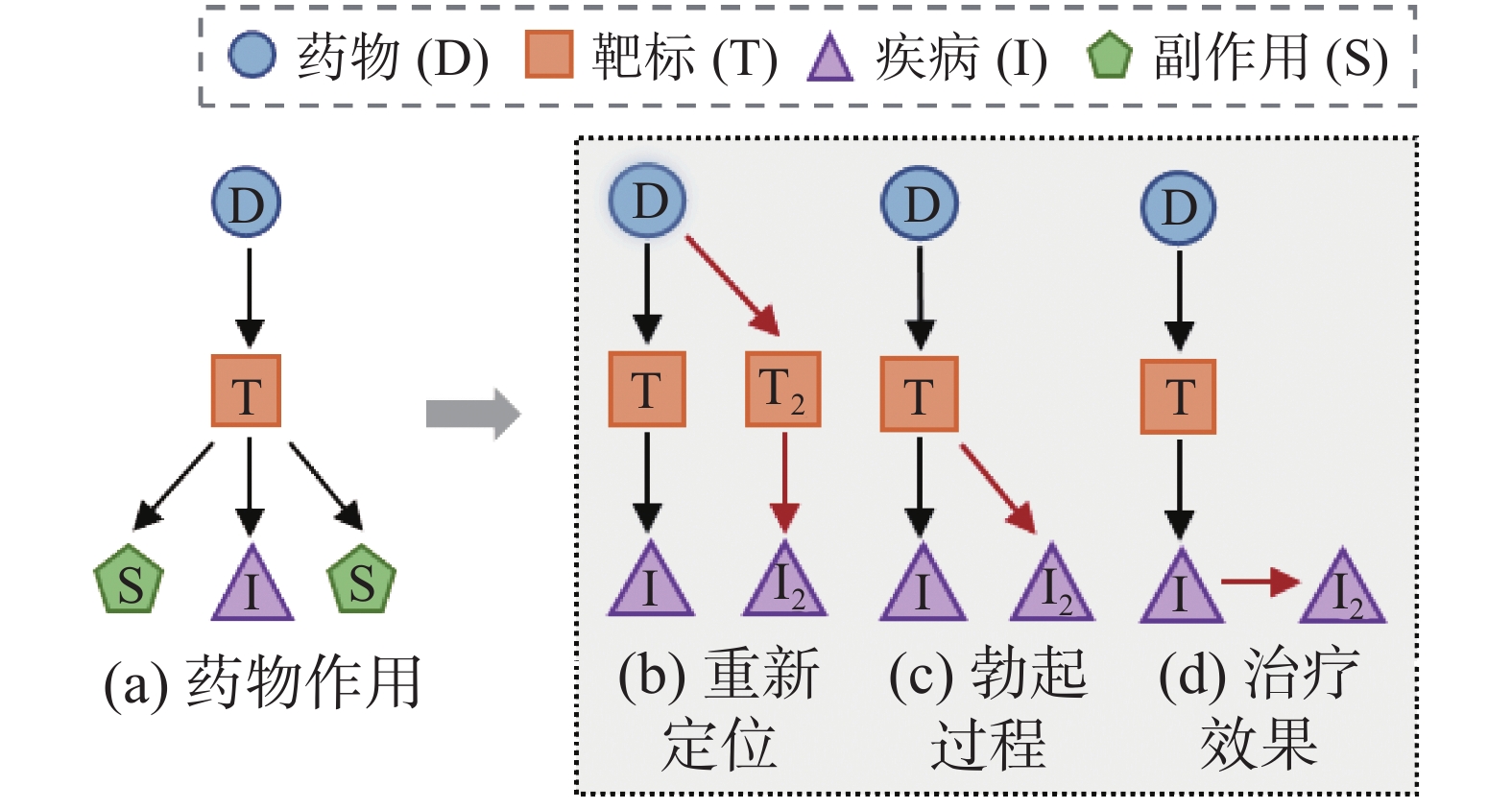 图 3 药物重定位依据Fig. 3 Opportunities of drug repositioning下载:
全尺寸图片
图 3 药物重定位依据Fig. 3 Opportunities of drug repositioning下载:
全尺寸图片
传统实验方法研究药物−靶标相互作用耗时耗资耗力且伴有一定的盲目性,难以进行大规模的药物−靶标相互作用识别工作。将人工智能技术尤其是深度学习技术运用到药物−靶标相互作用的挖掘、预测得到工业界和学术界的广泛青睐。目前,一些企业已成功将人工智能技术应用到分子活性预测[34]。例如,Atomwise公司运用深度学习算法预测药物−靶标相互作用亲和力,速度和准确性均处于领先地位。该公司的深度学习平台AtomNet包含超过160亿个用于虚拟筛选的分子。Exscientia公司开发了Centaur Chemist平台,利用大数据和AI针对特定靶标蛋白设计和筛选小分子化合物,为临床试验提供候选药物分子。2019年,Exscientia与GlaxoSmithKline公司合作,依托Centaur Chemist平台研发了治疗慢性阻塞性肺病的候选药物,大幅度提高药物研发效率。因此,研究基于新一代人工智能技术的药物−靶标相互作用预测不仅是国家层面的重点关注领域,也具有学术研究、现实应用意义和商业价值。
2. 研究现状
药物研发的首要步骤是识别与靶标结合会产生高亲和力的化学分子,从而进一步将化学分子优化为类药化合物(先导化合物)。由于对化学空间和蛋白质空间的动态关系了解有限,新药发现和靶标识别是一项充满挑战的任务。实验筛选先导化合物(如高通量筛选)耗费大量时间和金钱,而计算机辅助药物−靶标相互作用预测可以大幅度降低资源、时间和成本消耗,缩小候选药物筛选范围,降低物理实验筛选化学分子的需求。
分子对接是重要的计算机辅助药物设计技术,多种分子对接工具可供商业和学术研究使用,例如DOCK、AutoDock、FlexX、GOLD等[35-36]。分子对接包括两个步骤,将分子对接到靶标的结合位置(位置识别),然后预测对接构象与靶标的结合强度(打分)[28,37]。通过分子对接技术,可以清楚地了解药物与靶标结合的三维构象。但是,该类方法将药物小分子置于靶标蛋白的活性位置,通过不断地改变药物的构象识别出最优的药物−靶标结合构象,然后对对接构象与靶标的结合强度进行打分,因此需要在庞大的查找空间中遍历所有可能的构象以获得实际或接近于实际的药物−靶标结合构象,花费大量时间,消耗大量计算资源[27-28]。尽管目前有许多相对鲁棒且准确的结合位置识别算法,但打分函数的可靠性偏低[37],不同的分子对接算法预测的结合亲和力存在差异[32]。目前的方法将对接作为一个单独的过程,每一次对接都要重新开始,浪费时间和计算资源[28],且预测结果假阳性率较高[3]。有时仅仅一个氨基酸的不同就会完全改变结合位置的药理特性,这为自动分析和校正结构带来难题。
如图4所示,本文从使用的数据种类、问题定义、数据表示与编码、特征提取与特征融合、预测模块、学习方式等多角度多层次分析现有药物−标靶相互作用预测方法,从而帮助研究者更深入的地了解药物−靶标相互作用预测研究现状。
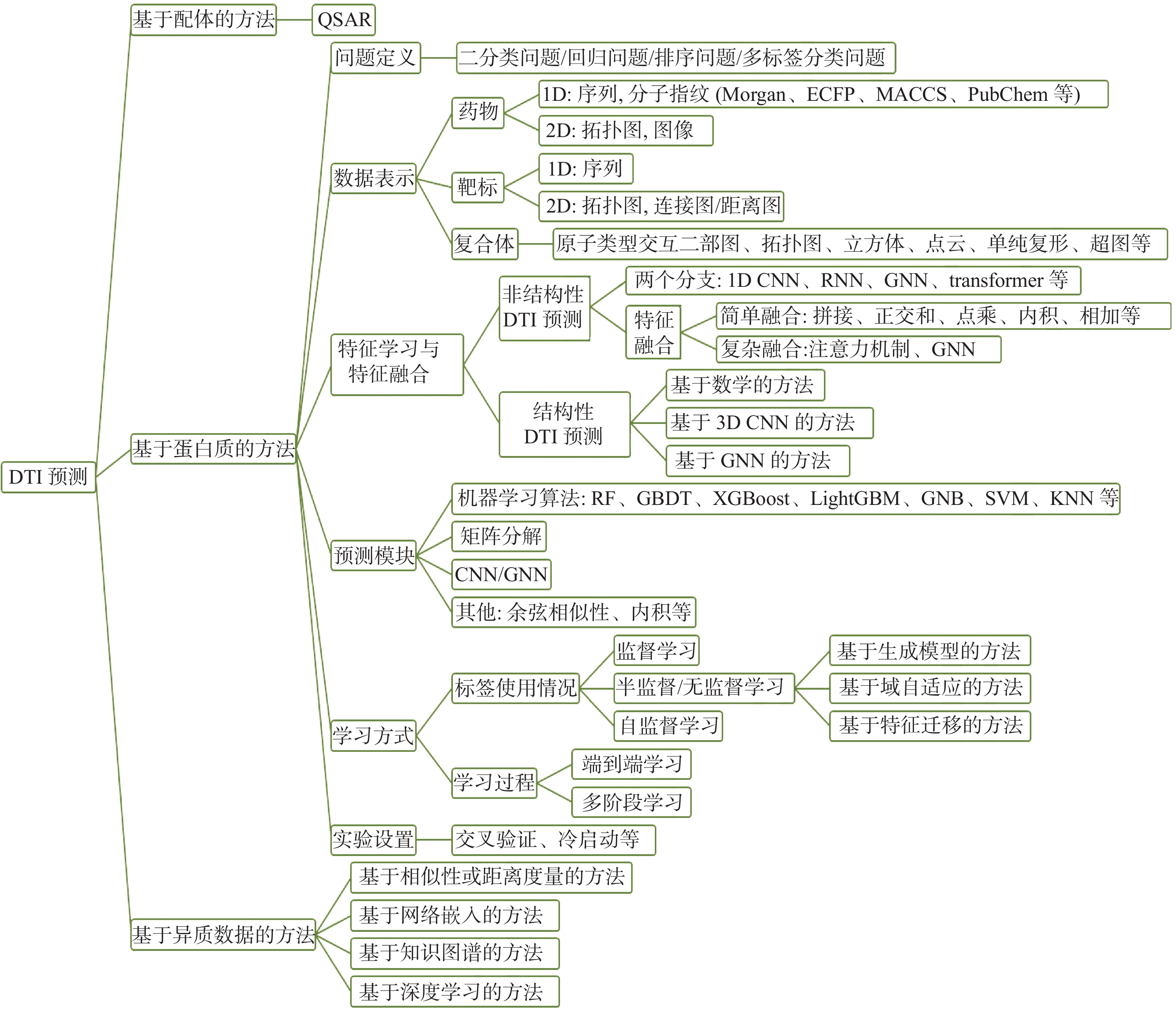 图 4 药物和靶标相互作用预测方法总结Fig. 4 Summary of drug-target interaction prediction下载:
全尺寸图片
图 4 药物和靶标相互作用预测方法总结Fig. 4 Summary of drug-target interaction prediction下载:
全尺寸图片
2.1 基于配体的方法
根据使用的数据不同,计算机辅助药物−靶标相互作用预测方法可以划分为基于配体的方法、基于蛋白质的方法和基于异质数据的方法。基于配体的方法只利用药物信息而不需要靶标信息,基于蛋白质的方法同时利用药物信息和靶标信息,基于异质数据的方法除了利用药物信息和靶标信息,还结合其他生物医学信息,如疾病、副作用等。基于配体的方法的主要假设是具有相似化学结构的药物会产生相似的生物活性,能与相似的靶标产生相互作用。使用聚类算法或分子指纹(fingerprint)相似性计算工具(如Tanimoto算法)可以得到药物分子之间的相似性。该类方法通过比较候选药物与靶标有已知相互作用关系药物的相似性预测候选药物与靶标是否会产生相互作用,而不依赖于靶标的任何信息。例如,Hu等[38]使用多任务学习方式联合学习不同靶标的配体特征。定量构效关系(quantitative structure-activity relationships,QSAR)是典型的基于配体的预测方法[34,39],根据配体结构和活性建立预测模型,以定量的形式来研究药物和靶标的相互作用。具体而言,QSAR通过对比候选药物与特定靶标的已知配体的相似性来评估候选药物和靶标的相互作用关系,可以在靶标结构未知的情况下,对分子进行有效筛选。其缺陷在于回归方程的物理意义模糊,无法帮助理解药物和靶标的作用机制。
基于配体的方法很直观,建立在广为接受的相似性原理上。但是在实际情况中,分子结构的微小改变都会导致迥然不同的生物结果。一些化合物在发生药理活性之前,它们的结构在细胞内会发生改变,因此,使用数据库中记录的化学分子结构在一定程度上会降低模型的预测结果。使用效力强的或优化后的化合物推断新的配体或训练模型,会产生预测结果对新的疾病药理性弱的风险。另外,对没有配体或已知配体数量不多的靶标,基于配体的方法预测结果较差。
2.2 基于蛋白质的方法
基于蛋白质的药物−靶标相互作用预测方法研究最为广泛,该类研究工作同时将药物分子数据和靶标分子数据作为模型输入[36-37,40-43,44-63]。模型的预测结果主要受到6个方面因素的影响。1)问题定义:药物−靶标相互作用预测可以定义为二分类问题或回归问题,前者预测药物和靶标是否会产生相互作用,后者预测药物和靶标的结合亲和力。2)数据表示:药物和靶标数据可以表示为一维、二维、三维或者图像的形式。3)特征学习与特征融合:输入数据的表示影响深度学习模型的选择,药物特征和靶标特征的融合方式影响药物−靶标相互作用预测结果。4)预测模块:预测模块的设计和选择也会影响药物−靶标相互作用预测结果。5)学习方式:模型可以以端到端或多阶段的学习方式预测药物−靶标相互作用。根据数据标签的使用情况,模型的学习方式还可以分为监督学习、半监督学习、自监督学习和无监督学习。6)实验设置:采用何种方式评价模型的预测效果,如交叉验证或者冷启动问题。此外,数据集的属性,包括数据集大小、数据分布、数据类型等,也会影响模型的预测结果。
2.2.1 问题定义
目前的研究工作主要将药物−靶标相互作用预测视为二分类任务或回归任务。二分类任务预测药物和靶标是否会产生相互作用,回归任务预测药物和靶标的结合亲和力得分。这两种任务对应药物研发中两个重要研究方向。分类任务预测药物和靶标之间有无关联,该任务的主要目的是为与疾病相关的生物靶标找到能够发生作用的靶向药物,或者为新的药物找到能够发生相互作用的生物靶标。回归任务主要目的是描述药物和靶标之间的相互作用机理,能够进一步反应药物的治疗效果。因此,药物−靶标结合亲和力预测任务是药物−靶标有无关联预测任务的更细粒度预测。这两种任务计算模型都能在很大程度上缩短药物研发时间,减少不必要的生物化学实验,从而实现高效的药物筛选。值得注意的是,当作为二分类任务时,由于缺乏可靠的负样本数据,许多重要的药物−靶标相互作用关系数据缺失,例如药物和靶标的剂量依赖性和定量亲和力,预测结果具有较高的假阳率。
此外,部分研究工作将药物−靶标相互作用预测作为排序问题[40-41]或多标签分类问题[42-43]。排序问题可以看作是特殊的回归问题。多标签分类问题是单标签分类问题的延伸,基于“多药物−多靶标”的药物研发范式,将输入特征向量映射到不同的标签,旨在利用不同标签之间的潜在相互关系。例如,有个药物和个靶标,在多标签分类任务中,将个药物(个靶标)作为输入样本,将个靶标(个药物)作为标签。多标签分类任务可以避免二分类任务将未知相互作用关系的药物−靶标对作为负样本而引入噪声的问题。但是,多标签分类任务的输出空间大小与标签数量呈指数关系(个标签会产生个标签集合),增加计算复杂度。为缓解此问题,研究者常采用聚类算法[42]、社区发现(community detection)算法[43]等将整个标签划分为多个子标签空间,然后在每个子空间运用多标签学习算法。
2.2.2 数据表示与编码
1) 药物数据表示
药物分子可以表示为一维字符串(string)、二维图(graph)或者图像。目前大多数的研究工作采用前两种药物分子表示方式,少部分研究工作根据SMILES序列生成二维图片,然后使用卷积神经网络(convolutional neural network,CNN)模型学习特征表示[64]。最常用的字符串是SMILES (simplified molecular input line-entry system)序列,用ASCII码表示分子结构。可以使用独热向量(one-hot vector)或多热向量(multi-hot vector)直接对SMILES进行编码,也可以用word2vec[65]将SMILES序列的字符映射为实值向量。此外,还可以使用词袋模型对SMILES进行编码,字典中的每个词表示SMILES序列的一种子结构[66]。同理,可以使用独热向量、word2vec编码子结构。SELFIES(self-referencing embedded strings)[67]是SMILES的改进版,解决了SMILES在句法上无效(无法对应一个化学分子图)或违反基本化学规则(如原子间最大价键数量)问题,适应于任何机器学习模型。在机器学习/深度学习模型中,常使用RDKit等软件包将SMILES序列转换为固定长度的分子指纹。分子指纹对分子中经常出现的子结构或功能团进行编码,表示为二进制向量或统计向量,向量中的元素表示子结构是否存在或者出现频次。根据指纹生成方式不同,分子指纹分为基于分子拓扑结构的指纹(例如Morgan、ECFP(extended-connectivity fingerprints))和基于SMARTS(SMILES arbitrary target specification)模式的指纹(例如MACCS、PubChem)。基于拓扑结构的指纹通过计算原子(键)的距离描述原子(键),而基于SMARTS的指纹根据SMARTS模式(预先定义的子结构模式词典)编码分子。药物分子的二维图描述了分子的拓扑结构特征和原子连接方式,图的节点是原子、边是键,节点的特征可以根据原子类型、邻接原子数量、邻接氢原子数量、原子隐性值、原子是否在芳香结构中等原子属性进行编码[68]。图5给出了药物数据常用表示形式。
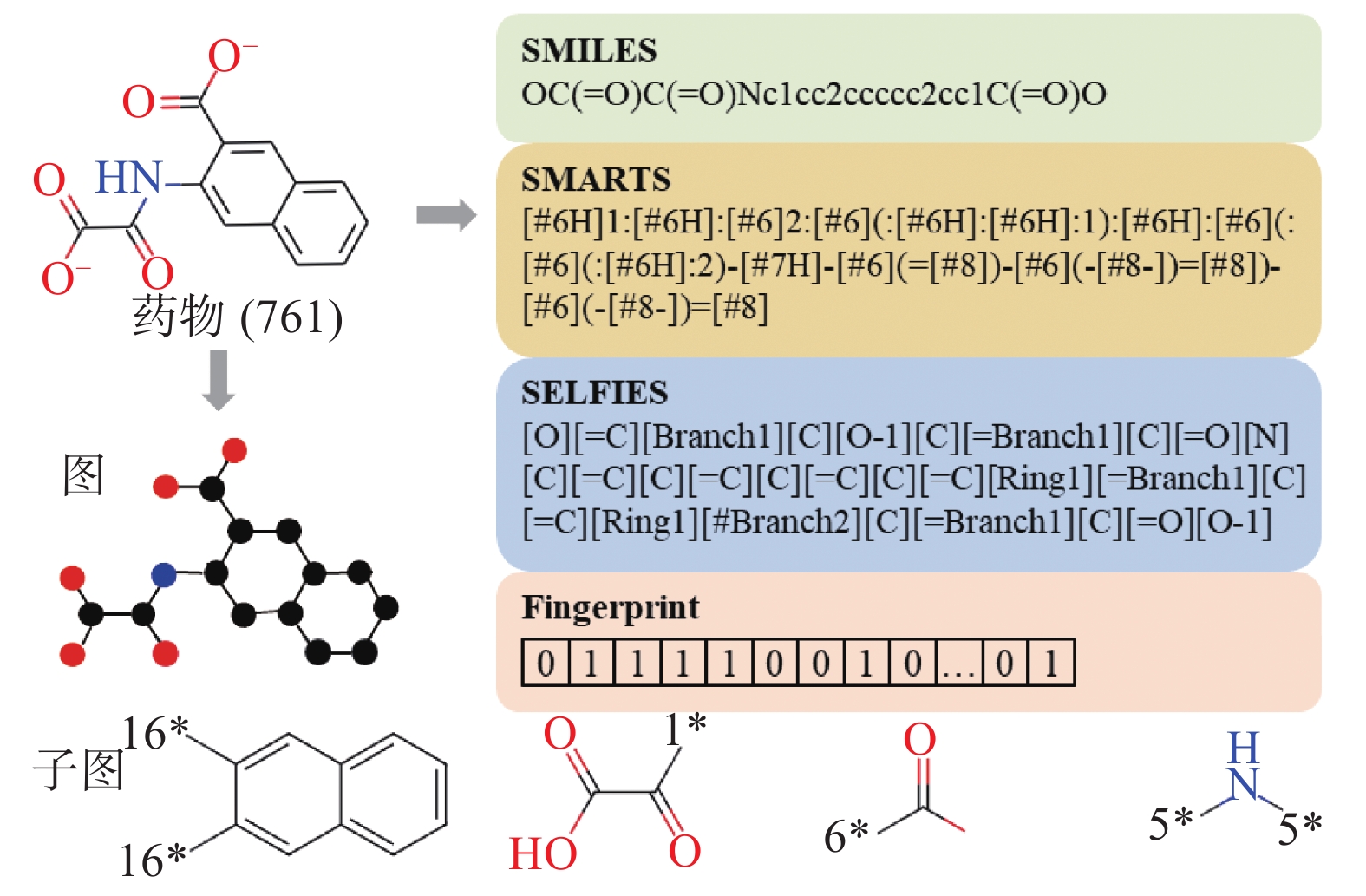 图 5 药物数据表示Fig. 5 Representations of drugs下载:
全尺寸图片
图 5 药物数据表示Fig. 5 Representations of drugs下载:
全尺寸图片
2) 靶标数据表示
靶标蛋白本质上是高度保留进化信息的氨基酸残基序列,用字母表示结构和物理化学属性。可以使用独热向量、word2vec、
$ \mathrm{k} $ -mer算法、SPS (structural property sequence)[69]、PSSM(position specific scoring matrix)[70]、PsePSSM(pseudo PSSM)[71]、PseAAC(pseudo amino acid composition)[72]等编码蛋白质序列,其中独热编码在深度学习模型中使用最为广泛。不同的靶标序列编码方式常结合在一起使用,以充分利用不同编码方式的优势。例如,We等[39]使PSC(protein sequence composition)描述子(由AAC(amino acid composition)、DC(dipeptide composition)和TC(tripeptide composition)组成)表示靶标,Mahmud等[73]使用PsePSSM、PseAAC和DC表示靶标。也可以将靶标蛋白表示为含有结构信息的二维连接图(contact map)或距离图。将靶标蛋白转换成带有拓扑结构信息的二维图也是一种常见的做法,图的节点是残基,邻接矩阵表示残基之间的距离。图6给出了靶标数据常用表示形式。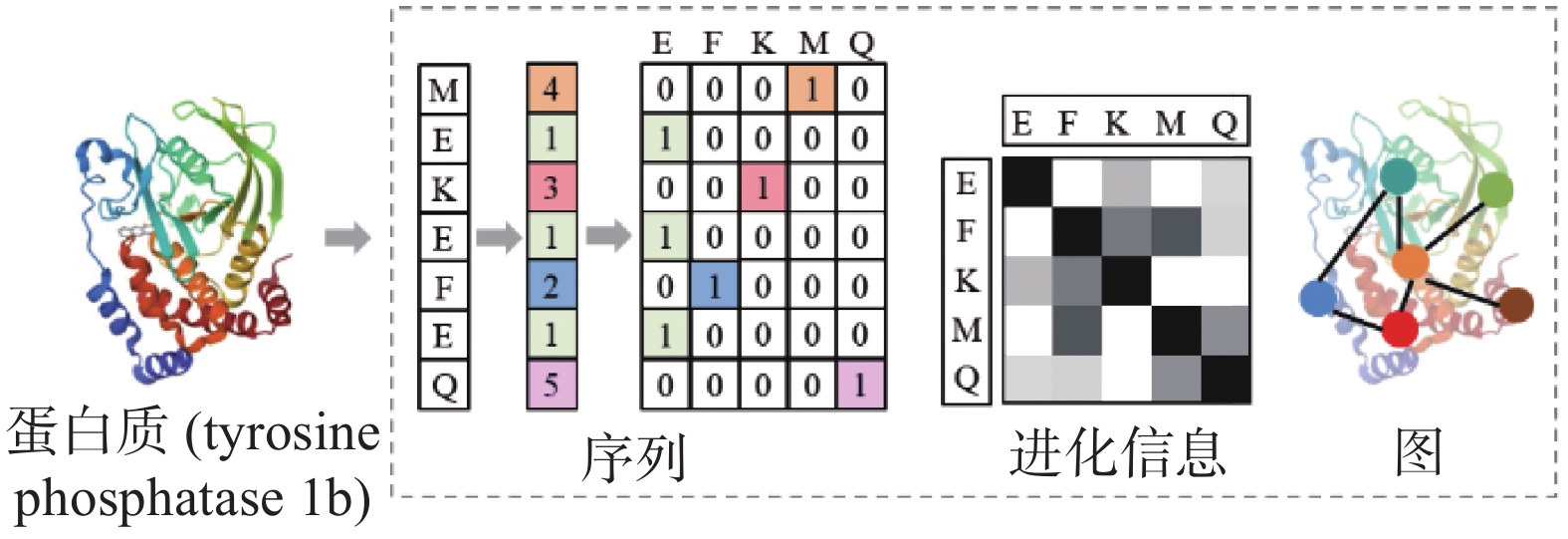 图 6 靶标数据表示Fig. 6 Representations of targets下载:
全尺寸图片
图 6 靶标数据表示Fig. 6 Representations of targets下载:
全尺寸图片
另外,蛋白质三维结构包含更多氨基酸序列空间组织信息,与其相应的生物功能和靶标结合特性有着更直接的关联。基于三维结构的药物−标靶相互作用预测也得到广泛关注。在三维空间中,药物−靶标复合体以
$ x $ 、$ y $ 、$ {\textit{z}} $ 坐标进行观察,每一个药物分子原子和靶标分子原子都有一个三维坐标信息。药物和靶标原子类型关系二部图/矩阵(如C-N、O-O二部图/矩阵)[36-37,44-46]、三维立方体[47-52]、三维点云[53]、图[54-63]、单纯复形(simplicial complex)[74-75]、超图[76]等常用来建模药物−靶标复合体的三维结构信息或药物和靶标分子间的相互作用关系。将药物−靶标复合体表示为图时,图的节点是药物或靶标的原子,边表示原子之间的共价键或非共价键关系,非共价键关系根据原子之间的距离得到。根据药物−靶标复合体的表示形式,可以选用不同的深度学习模型学习复合体的结构特征,也可以采用基于图论、几何学、拓扑学等的算法提取药物−靶标复合体的特征表示。此外,在图神经网络(graph neural network,GNN)模型中,原子的三维坐标信息常转换为原子的相对空间结构信息结合在GNN模型的消息传递和聚合过程,如原子之间的距离、键之间的夹角、平面角等。2.2.3 特征提取与特征融合
特征提取是药物−靶标相互作用预测的关键步骤。特征提取的目的是从原始数据中挖掘信息丰富、具有判别性和非冗余的知识,进而可以更好的用于下游任务或进行的接下来的步骤。特征提取的方式分为以数据驱动和不以数据驱动两种,前者自动的从原始数据中学习特征表示,后者依赖特征工程,使用数学工具或根据预预先定义的规则计算得到特征表示,是一种手工提取特征(hand-crafted feature)。基于机器学习的方法不以数据驱动,将手工提取特征(如描述子(靶标的PseAAC、药物的DC等)、相似性、统计信息、特征值、Forman-Ricci曲率等)输入至机器学习模型得到预测结果[17,40-41,45,73-74,77-87]。基于机器学习的方法的缺陷在于预测结果在很大程度上依赖于提取的特征的质量,在大规模数据集上泛化能力较差。基于深度学习的方法以数据为驱动,直接从药物/靶标的原始数据(序列、图等)或描述子中学习特征表示和数据分布,预测结果准确性更高,模型的泛化性更好。
根据预测模型是否考虑药物和靶标的三维结构信息,药物−靶标相互作用预测方法分为结构性药物−靶标相互作用预测和非结构性药物−靶标相互作用预测,目前大多数的研究工作主要关注后者。非结构性预测方法不考虑药物−靶标复合体的三维结构信息,只利用药物和靶标的序列或二维拓扑结构信息。这类方法通常将预测模型设计为包含药物编码器和靶标编码器的两个分支,分别用于学习药物特征表示和靶标特征表示。根据药物数据和靶标数据的表示方式不同选用不同的深度学习模型或者模型组合。例如,当药物表示为一维序列时,可以采用1D CNN、循环神经网络(recurrent neural network,RNN)、transformer[88]或者两种及两种以上的模型组合编码药物特征表示。当药物表示为二维图时,可以使用GNN模型编码节点特征,然后经过池化操作得到药物的特征表示。学习到的药物特征和靶标特征通过拼接[69,73,89-94]、正交和[77]、点乘[66,95-97]、内积[98-100]、相加[101]等方式进行特征融合得到药物−靶标对的特征表示,其中拼接是最常用的特征融合方式。
采用两个独立分支分别编码药物特征和靶标特征忽略了药物和靶标之间的相互作用关系。许多研究工作利用注意力机制或图神经网络编码药物和靶标之间的相互作用关系,从而更好地融合药物特征和靶标特征。基于注意力机制的方法编码药物编码器和靶标编码器不同阶段(输出层、中间层)的输出特征之间的相互作用强度。如使用注意力机制编码药物特征和靶标特征的作用强度[102-104]、药物和靶标在“字母”级别的作用强度(药物原子或子序列、靶标残基或子序列)[69,105-115]、药物与靶标子序列或药物子结构与靶标之间的作用强度[116-119]。基于图神经网络的方法建模药物分子原子和靶标分子原子之间、药物和靶标实体之间、或药物−靶标对之间的交互关系[95,120-122]。例如,Nguye等[120]提出一种graph-in-graph网络结构建模药物和靶标蛋白残基之间的关系,即将药物表示为二维图,同时将药物和靶标残基作为节点,将靶标残基之间的连接图和药物与靶标残基之间的关系作为边构建另一个图。MVGCN[123]根据药物−药物相似性、靶标−靶标相似性以及药物−靶标相互作用关系构建图。除了用两个分支编码药物和靶标的特征外,HGRL-DTA[121]还引入第3个分支用GNN编码药物−靶标二部图。GCN-DTI[122]将药物−靶标对作为节点,建模药物−靶标对之间的关系。
非结构性药物−靶标相互作用预测方法不需要靶标蛋白的三维空间结构信息,可以适用于一些三维结构无法获得的靶标蛋白,如G蛋白偶联受体。但是,这类预测方法无法准确描述药物分子原子和靶标分子原子在三维空间复杂的作用关系。具有相同二维拓扑结构图的分子常常具有不同的三维分子结构,并且具有不同的分子属性[124]。研究表明蛋白质通过特定的三维构体(conformation)发挥生物功能[125],分子的相互作用受构体中的原子影响[126]。因此,有效的建模药物−靶标复合体的三维结构可以进一步提高药物−靶标预测的准确性。此外,随着生物信息技术的快速发展,如AlphaFold II[16],准确的药物−靶标复合体位置预测等,越来越多的生物结构数据可以获得,为结构性药物−靶标相互作用预测研究提供了机会。
结构性药物−靶标相互作用预测方法根据药物−靶标复合体的三维结构预测药物和靶标的作用关系,面临的主要挑战是建模药物分子和靶标分子之间的相互作用关系和药物−靶标复合体的三维空间结构。目前这类研究工作主要分为基于数学的方法,基于3D CNN的方法和基于GNN的方法。基于数学的方法使用预定义规则从药物和靶标的原子类型组合关系中提取描述子(例如原子类型组合出现的频次、子结构出现的频次)[37,44-46,127],或者使用基于几何、拓扑结构、图论的算法提取描述子(如Forman-Ricci曲率,特征值等)[17,74-75,87,128-130]。提取的描述子通常与机器学习模型(例如RF(random forest)[131]、GBT(gradient boosting tree)[132])或深度学习模型(如CNN、GNN)相结合预测药物−靶标的相互作用。例如,PerSpect[75]提出基于距离的原子类型交互矩阵和基于静电的原子类型交互矩阵。基于距离的原子类型交互矩阵考虑4种类型药物分子原子(C、N、O、S)和9种类型蛋白质原子(C、N、O、S、P、F、Cl、Br、I)的组合关系,共36种原子类型组合。基于静电的原子类型交互矩阵还会考虑药物分子和蛋白质中的H原子,共50种原子类型组合。PerSpect采用一种过滤(filtration)策略,设置多个距离阈值,将每一距离阈值内的每种原子类型组合关系表示为单纯复形,计算单纯复形的特征值作为描述子。对计算得到的特征值进一步操作(如求均值、最大值、最小值等)便可以得到药物−靶标复合体的特征表示,然后使用GBT得到预测结果。类似的,FPRC[74]计算单纯复形的Forman-Ricci曲率作为描述子。基于PerSpect,Mol-PSI[130]在多个尺度上将非零特征值转换为二维图像(
$ x $ 轴表示特征值,$ y $ 轴表示多尺度参数),同时将零特征值转换为一维图像,然后采用模型集成策略结合多尺度和多原子类型组合表示提高药物−靶标相互作用预测的准确性。基于数学的方法非常依赖特征工程,在大规模数据集上泛化能力较差。此外,基于数学的方法由于只考虑药物和靶标之间的原子类型交互关系,忽视了药物或靶标内的原子相互作用关系以及多种类型原子的局部关系,因此这类方法只能提取有限的药物−靶标复合体中的空间结构信息。基于3D CNN的方法将药物−靶标复合体转换为规则的三维立方体,然后用3D CNN模型学习复合体的特征表示用于药物−靶标相互作用预测[47-52]。基于3D CNN的方法存在以下缺陷:一方面,基于3D CNN的方法忽略了药物−靶标复合体的拓扑结构信息;另一方面,虽然三维立方体可以描述药物−靶标复合体的局部空间结构信息,但由于药物−靶标复合体中的原子并不是规则排列的,构建的三维立方体中含有大量的空值,稀疏性高,占用计算内存,且计算效率低。此外,构建的三维立方体不具有旋转不变性,也无法描述原子之间的距离信息。
基于GNN的方法得益于GNN模型优异的图结构数据建模能力[133],又可以进一步划分为基于拓扑结构的方法和基于空间结构的方法。基于拓扑结构的方法可以看作是非结构性药物−靶标相互作用预测方法在结构性数据上的延伸,该类方法保留药物、靶标或复合体的拓扑结构信息[35,44,47,54-56,59-61,130],而不考虑原子间的相对空间结构信息,如原子间距离、键的夹角、二面角等。例如PLIG[18]保留药物分子的拓扑结构,即构建以药物分子原子为节点、键为边的图,节点初始特征除了原子自身的属性外(如原子类型),还包含与之距离在预定义阈值之内的靶标蛋白分子中原子的信息。靶标表示成序列用1D CNN学习,或者不用靶标的信息。一些研究工作构建药物−靶标复合体关系图,图的节点是药物分子原子或靶标分子原子,边表示原子间的共价键或非共价键关系[47,59-60]。还有一类研究工作构建3个图分别表示药物、靶标以及药物分子原子和靶标分子原子作用关系[55-56,61]。学习到的药物、靶标、复合体的特征,可以采用前面提到的特征融合方式进行融合,如拼接[55]、注意力机制[54]等。基于拓扑结构的方法的缺陷是无法区分具有相同拓扑结构但几何结构不同的分子如trans-1、2-dichloroethene和cis-1、2-dichloroethene。
基于空间结构的方法根据药物−靶标复合体的拓扑结构,以及相对空间信息编码药物−靶标复合体的特征表示。相对空间信息包括距离[57,63,134-136]、角度[58]等,根据原子的三维坐标计算得到。例如MP-GNN[136]将每种药物和靶标的原子类型交互关系表示为二部图,节点的特征仅与原子间的距离有关。Zhou等[57]将药物−靶标复合体表示为图,图的节点表示药物分子原子和靶标分子原子,邻接矩阵根据节点之间的距离构建,并编码距离信息融入到节点和边(节点对)的消息传递与聚合过程。类似地,SS-GNN[62]也将药物−靶标复合体表示为图,但它使用GNN模型和多层感知机(multilayer perceptron,MLP)分别编码节点特征和距离特征,最后将两种特征根据边用拼接的方式进行融合。GLI[63]根据拓扑结构和距离信息构建药物图、靶标图和药物−靶标复合体相互作用图,然后从局部和全局两个角度学习复合体的特征表示。GLI先用GNN模型分别从药物图和靶标图中学习节点特征,在全局视角方面,GLI将药物和靶标作为节点,预测药物−靶标长距离作用关系;在局部视角方面,GLI使用注意力机制聚合靶标节点特征,权重系数由原子间距离决定,基本思想是距离药物越近的靶标原子对相互作用贡献越大。但是仅仅使用距离信息并不能充分的描述药物−靶标的三维结构信息。原则上,所有原子间的距离矩阵包含药物−靶标复合体的全部几何信息。但是,将整个距离矩阵引入到GNN的消息传递过程会大大增加计算复杂度,且容易导致过拟合。因此,基于GNN的方法通常以药物为中心,只保留与药物原子距离在预定义阈值之内的靶标原子,但是这样会使GNN模型无法区分某些分子。例如,当阈值为2
$ Å $ 时,GNN无法区分具有相同的键长的六边形和含有两个三角形的分子(如Cyclohexane和Cyclopropane),因为这两个分子的每一个原子具有相同的邻域节点。除了原子之间的距离信息,SIGN[58]进一步考虑角度信息,将角度划分为若干个域,在每个域内聚合一定距离内的节点信息。另外,GNN模型通常只包含两三层,过多的层数会产生过平滑问题,因此每一个原子不能从长距离原子聚合信息。键的拓扑结构为描述药物−靶标复合体空间信息提供了一个新的角度,可以挖掘高阶原子信息,有利于复合体的特学习。鉴于此,Yi等[134]构建以键为节点的图建模药物−靶标复合体全局信息。基于空间结构的方法因为在原子级别上建模药物−靶标复合体的三维空间结构信息,计算量大且计算复杂度高。未来,结构性药物−靶标相互作用预测有待进一步探索。总的来说,各种方法并不是完全独立的,许多研究工作会同时考虑药物和靶标不同的特征表示,采用混合模型或模型集成算法学习并融合不同特征的互补信息,提高药物靶标相互作用预测准确性[48,79,86,94,97,100,137-145]。例如,Jones等[143]同时将药物−靶标复合体表示为三维立方体和图(节点表示药物分子原子和靶标分子原子,边表示原子之间的共价键和非共价键关系),分别用3D CN模型和GNN模型学习复合体的特征表示。
2.2.4 预测模块
预测模块的选择也会影响药物−靶标相互作用的预测结果。机器学习模型/算法、MLP、矩阵分解、GNN、CNN等常作为预测模块,其中,MLP应用最为广泛。机器学习模型/算法包括RF、GBT、XGBoost(extreme gradient boosting)[146]、LightGBM[147]、GNB(gaussian naive bayes)[148]、KNN(k-nearest neighbor)、逻辑回归(logistic regression)、SVM(support vector machine)[149]、KronRLS[40]等,既可以根据手工提取特征预测药物−靶标的相互作用[40-41,45,73,77-84],也可以与深度学习模型相结合,利用深度学习模型提取的特征预测药物−靶标相互作用[137,141,150]。基于矩阵分解的方法的假设是药物和靶标处于相同的距离空间,那么药物和靶标之间的距离可以用来衡量它们之间相互作用的强度[151-158]。因此,在一定约束条件下,药物和靶标都可以嵌入到共同低维子空间。但是,基于矩阵运算的计算模型需要消耗大量的计算资源,随着药物数据和靶标数据数量和多样性急剧增加,该类方法不再适用,而且该类方法不易于推广于新的药物−靶标相互作用关系预测。一些研究工作也会采用CNN[66,159-160]、GNN[95,161]作为预测模块,将GNN作为预测模块的主要目的是挖掘药物−靶标对的高阶关系。此外,提取的药物特征和靶标特征的余弦相似性[140,162]、内积[98-100,163]等也可以直接作为预测结果。图7以药物−靶标复合体PDB ID 1c84(药物名称761,蛋白质名称tyrosine phosphatase 1b)为例给出了药物−靶标相互作用预测流程。
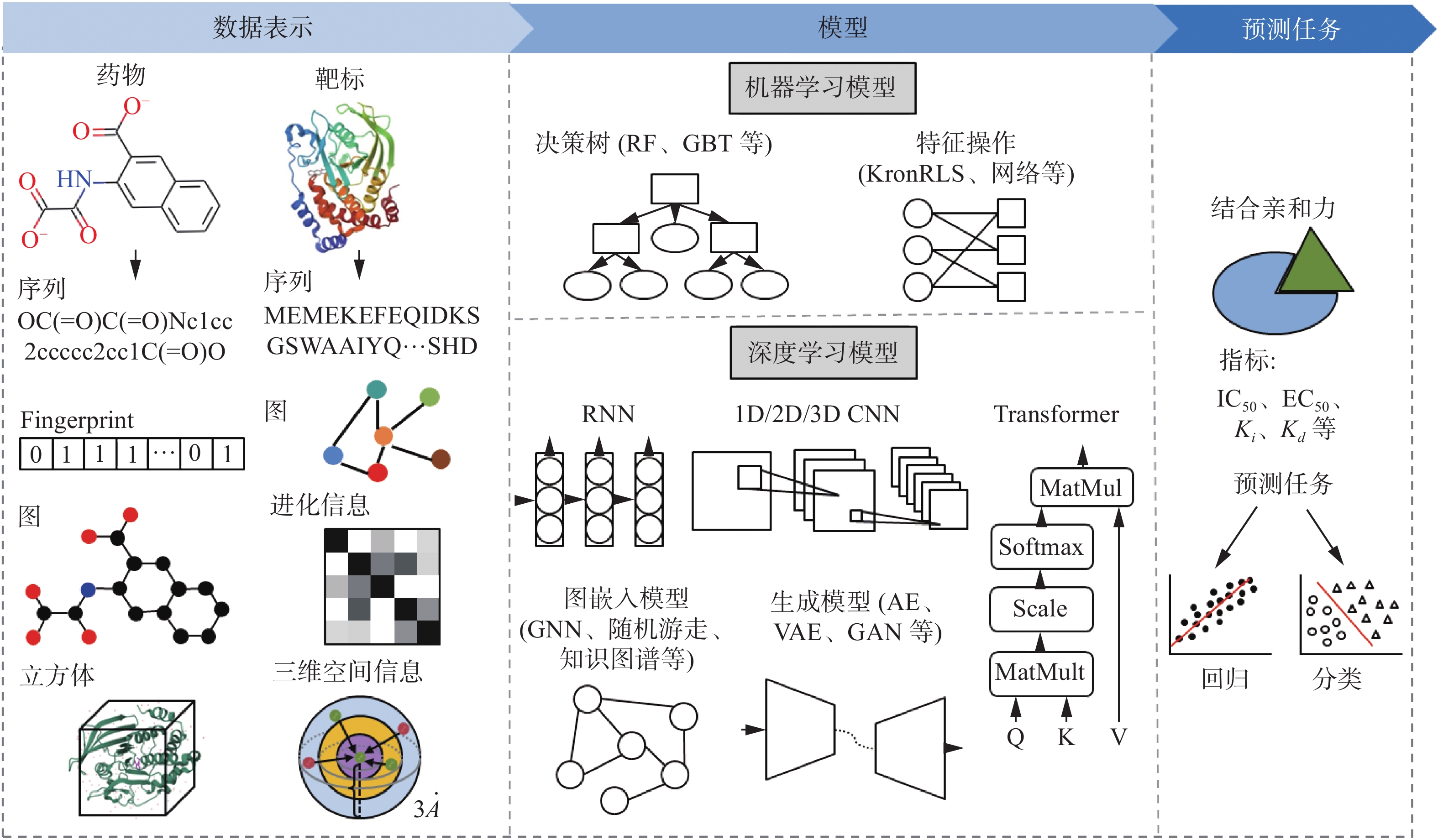 图 7 药物−靶标相互作用预测流程Fig. 7 Flowchart of drug-target interaction prediction下载:
全尺寸图片
图 7 药物−靶标相互作用预测流程Fig. 7 Flowchart of drug-target interaction prediction下载:
全尺寸图片
2.2.5 学习方式
根据数据标签的使用情况,药物−靶标相互作用预测模型的学习方式可以划分为监督学习、半监督学习、无监督学习和自监督学习。大多数的药物−靶标相互作用预测方法采用监督学习方式,每一个输入样本对应一个标签(0/1,或亲和力得分),预测模型的参数根据损失函数使用反向梯度算法优化。当将药物−靶标相互作用预测作为回归任务时,使用均方误差(mean squared error,MSE)(又称L2损失函数)或均绝对值误差(mean absolute error,MAE)(又称L1损失函数)作为损失函数,衡量预测亲和力与实际亲和力之间的偏差;当将药物−靶标相互作用预测作为二分类任务时,既可以使用二值交叉熵(binary cross-entropy)损失函数,也可以使用均方误差损失函数,此时预测结果是归一化0−1的连续值。
不同于自然语言处理和计算机视觉领域有大量的训练数据,如广泛使用的ImageNet数据集ILSVRC(ImageNet large scale visual recognition chall-enge)2012-2017(https://www.image-net.org/download.php)训练集包含120多万张图像,已知具有相互作用关系的药物−靶标样本只是冰山一角,而且获得药物和靶标之间的相互作用关系需要进行湿实验验证,耗时耗资耗力。另外,由于实验平台、使用剂量、批次效应等因素的影响,数据集会有噪声,不同数据集会存在分布偏移(distribution shift)问题。监督学习方式依赖样本标注,只能学习数据集本身的分布,且预测结果偏向于具有相互作用关系较多的药物和靶标,在数据分布不同的数据集上泛化性能较差。半监督学习、无监督学习和自监督学习可以缓解模型对标签样本的过度依赖,提高模型的预测结果和泛化能力。
基于半监督/无监督学习的药物−靶标相互作用预测方法可以划分为基于生成模型的方法、基于域自适应的方法和基于特征迁移的方法。基于生成模型的方法旨在增强预测模型的鲁棒性和泛化性能,常用的生成模型包括自编码器(auto-encoder,AE)、变分自编码器(variational auto-encoder,VAE)和生成对抗网络(generative adversarial network,GAN)。大部分研究工作使用大量无标签药物/靶标数据预训练自编码器[69,140,164-166],然后将预训练得到的药物/靶标编码器微调至药物−靶标相互作用任务。例如,Graph-CNN[166]、Co-VAE[167]和GANDTI[150]使用变分自编码器学习药物/靶标的隐藏特征表示,GANsDTA[159]使用GAN生成假的药物序列和靶标序列,增加训练样本的数量和多样性。基于生成模型的方式需要对原始数据进行重构,重构计算开销大,且GAN的训练是一件充满挑战的任务,因而增加了任务的复杂度。
基于域自适应的方法先用训练集/源域数据学习训练集/源域预测模型,然后使用对抗学习策略、知识蒸馏算法等学习测试集/目标域的预测模型,旨在提高模型在测试集/目标域的预测结果和模型的泛化的能力[105,109]。基于特征迁移的方法主要得益于近年来大语言模型(如ChatGPT-4[168])在许多自然语言任务上都取得最先进的结果。药物和靶标也可以看成是一种特殊的自然语言,因此,一些研究工作也将预训练语言模型应用到药物−靶标相互作用预测任务。例如,利用预训练语言模型从大量无标签药物序列和/或蛋白质序列中学习药物/靶标的子序列初始特征[66,169]、氨基酸残基的初始特征[120]、药物/靶标的初始特征[112,170]等。近年来,化学分子预训练模型和蛋白质预训练模型也纷纷涌现。GraphMVP[171]从化学分子二维图、三维图两个视角预训练模型,并用于药物−靶标相互作用预测任务。综述[172]详细总结了各种化学分子预训练模型。ESM-1b[173]基于Transformer[88],是蛋白质预训练模型,共33层,650 000 000个参数,使用250 000 000个蛋白质序列预训练得到。此外,一些研究工作将从相似任务和相关任务学习到的药物特征表示和靶标特征表示迁移至药物−靶标相互作用预测任务。例如GraMDTA[144]利用异质数据缓解数据稀疏问题,在异质图上预训练GNN模型学习药物和靶标的特征表示。Nguyen等[170]预训练药物−药物相互作用、和靶标−靶标相互作用预测模型,将预训练的药物编码器和靶标编码器迁移到药物−靶标相互作用预测任务。
自监督学习主要是利用辅助任务(pretext)从数据中挖掘自身的监督信息,通过这种构造的监督信息对网络进行训练,从而可以学习到对下游任务有价值的表征。例如,SIGN[58]和ELGN[156]将重构药物分子原子和靶标分子原子之间的远距离关系作为辅助任务。对比学习是最常用的自监督学习方法,基本做法是构建正负样本对,核心思想是使相似的样本在特征空间距离更近,同时使相似度低的样本在特征空间距离更远。常用的负样本生成策略包括扰动节点特征(如节点级别洗牌或特征级别洗牌)、扰动邻接矩阵(如增减边)、掩藏节点(node masking)等[98,101,123,154]。在实际操作过程中,除了对正负样本的特征使用对比损失函数外,对从两个角度提取的特征运用对比损失函数也是一种常用的策略。例如,SGCL-DTI[174]根据药物−靶标样本的连接关系(是否共享相同的药物和靶标)和特征相似性构建以药物−靶标对为节点的图,并对这两个角度学习的特征使用对比损失函数。SupDTI[142]扰动节点特征生成负样本,在正负样本节点特征之间、节点特征和图特征之间两个尺度上使用对比损失函数。前面提到的基于生成模型的药物−靶标相互作用预测方法也可以归为基于自监督学习的方法。
另外,根据预测模型的特征提取和结果预测是否是分开独立进行的,预测模型的学习方式又可以进一步划分为端到端学习和多阶段学习。端到端学习方式直接从输入数据得到预测结果,而多阶段学习方式先提取药物和靶标的特征表示,再进行相互作用预测。目前,大多数的深度学习模型以端到端的方式预测药物−靶标相互作用,与涉及特征计算和特征选择的模型相比,端到端模型需要大量的数据来学习药物和靶标之间的复杂关系。多阶段学习的特征提取方式因为与目标无关,可以用于不同的下游任务,这也意味着提取的特征会偏离目标下游任务,预测结果准确性低且不稳定。
2.2.6 实验设置
药物−靶标相互作用预测可以划分为4种不同的实验设置:1)热启动——训练集中包含测试集中的药物分子和蛋白质分子;2)药物冷启动——训练集中不包含测试集中的药物分子,但包含测试集中的蛋白质分子;3)靶标冷启动——训练集中不包含测试集中的蛋白质分子,但包含测试集中的药物分子;4)药物和靶标冷启动——训练集中既不包括测试集中的药物分子也不包括测试集中的蛋白质分子。目前大多数的研究工作主要采用交叉验证方式评估模型预测结果,关注的主要是第一种实验设置,训练得到的模型在预测新药物和新靶标相互作用关系上泛化能力较差。实际上,大多数药物(靶标)的已知相互作用关系都较少,而较少的数据量不足以支撑学习到的模型具有较强的鲁棒性和较高的预测准确性,且容易使模型过拟合。大部分预测模型关注模型在测试集上的整体预测结果,牺牲的是模型在具有较少相互作用关系的药物(靶标)上的预测性能。Li等[175]将预测以一个药物为核心结合不同靶标的相互作用关系或预测以一个靶标为核心结合不同药物的相互自作用关系作为一个任务,采用元学习策略解决药物−靶标相互作用预测的冷启动问题,旨在提高模型的泛化性能和模型预测新药物和新靶标相互作用关系的准确性。
另外,训练集中药物分子(蛋白质分子)和测试集中药物分子(蛋白质分子)之间的高相似度也会造成对模型预测结果的过高评估。为进一步分析模型的鲁棒性和泛化性,许多研究工作[158,161,174,176]会去除训练集中与测试集中药物或靶标相似度在某一阈值之上的药物−靶标相互作用样本。
2.3 基于异质数据的方法
药物可以发挥多种功能,产生多种与其药理学有关的作用方式(mode of action),这为药物重定位带来机遇[19]。通过观察特定环境中的基因表达可以了解生物系统。根据生物系统的状态,从转录的mRNA的数量可以判断某些基因是否过度表达或表达不足。基因的非正常表达可以视为化学分子反应指标(proxy),称为基因表达标记(gene expression signature)。其中,CMap(connectivity map)[177]已成功用于药物重定位,其核心思想是从基因谱中可以挖掘和对比药物的作用方式。此外,mRNA的表达不仅可以反应药物活性也可以用来表征疾病状态。疾病通常根据病理(如感染)或观察到的生物功能障碍(如细胞不受控制生长)进行分类。相似的疾病治疗方式相似,因此疾病关联网络可以为药物重定位提供有价值的信息。表型(phenotype)是一个有机体或生物体的一组特征或特点,如形态学、发展、生物化学或物理特点等。基于表型的药物筛选贯穿药物研发的整个过程,是最有效的新药投入市场方式[19]。副作用(side-effect)是表型的一种,西地那非的例子很好地说明了表型对药物重定位的重要性。半个多世纪前,基于表型的方法是药物研发的核心。随着分子生物学的到来,现在主要是基于蛋白质的方法。图8给出了不同生物医学概念(biomedical concept)关联图,黄色表示与药物和药物作用方式相关的联系(虚线表示药物与RNA通过CMap建立联系),蓝色表示生物概念,灰色表示疾病,红色表示药物可用于的适应症/疾病。
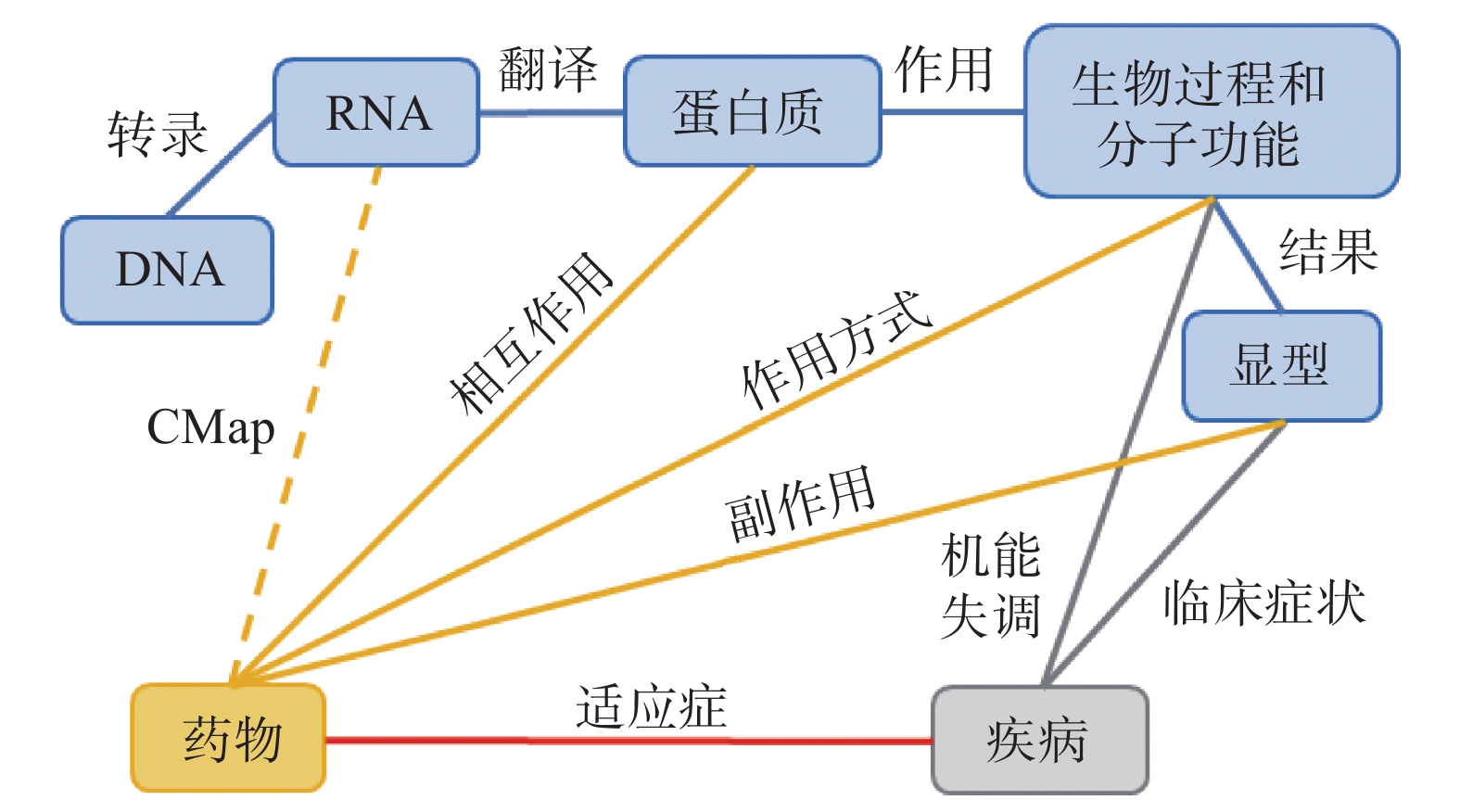 图 8 不同生物医学概念关系Fig. 8 Relationships of different biomedical concepts下载:
全尺寸图片
图 8 不同生物医学概念关系Fig. 8 Relationships of different biomedical concepts下载:
全尺寸图片
随着高通量测序成本不断降低,大规模并行技术以及新的传感技术的发展,产生了大量以药物为核心的数据,包括化学分子结构(SMILES)、ATC(anatomical therapeutic chemical)编码(由WHO成立,将药物划分为5个等级,每一个等级下又划分包括若干个子类别)、副作用、GO(gene ontology)术语、疾病以及生物医学实体之间复杂的联系,这些异质数据为描述药物和靶标之间的相互作用关系提供了多视角信息。融合多种类型的数据可以弥补单一类型数据缺失或者信息不准确的问题,降低预测结果的假阳性[178]。生物医学实体之间的复杂关系既可以表示为多个与药物相关的网络和与靶标相关的网络,也可以表示为一个生物医学异质图,图的节点表示生物医学实体(如药物、靶标、疾病、副作用),图的边表示实体之间的关系(如靶标−靶标相互联系,靶标−疾病关系)。药物−靶标相互作用预测可以看成链路预测问题,即一个药物节点和一个靶标节点之间是否存在联系。现有的基于异质数据的药物−靶标相互作用预测方法可以划分为基于相似性或距离度量的方法、基于随机游走的方法、基于知识图谱的方法和基于深度学习的方法。
2.3.1 基于相似性或距离度量的方法
基于相似性或距离度量的方法是应用最多的一类药物−靶标相互作用预测方法,其基本假设是相似的药物会与相似的靶标产生相互作用,根据相似性可以推断出脱靶蛋白。该类方法根据药物的化学分子结构或与药物相关的信息,使用相似性函数或距离函数计算药物之间的相似性。同理,根据靶标蛋白序列或与靶标相关的信息可以得到靶标之间的相似性。得到的药物−药物(靶标−靶标)相似性矩阵既可以直接作为药物(靶标)的特征表示,也可以输入至CNN、AE等深度学习模型学习药物(靶标)的深度特征表示,然后由预测模块预测药物−靶标对之间的相互作用关系[25,27,79,85-86,150,155-156,158,164,179-188]。基于相似性的方法常与随机游走(random walk with restart,RWR)[139,150,156,164,185,188-189]、矩阵分解方法[79,85,156,158,179,181,186]、核函数方法[179-180,182]等结合。
两个药物分子结构的相似度常用Tanimoto系数、Jaccard相似系数、欧氏距离(Euclidean distance)、余弦相似性(cosine similarity)等衡量。靶标序列之间的相似度用归一化的Smith-Waterman算法衡量。药物(靶标)与某类实体的关系可以描述为一个二部图,用矩阵表示,矩阵的每一行都是一个二值向量,表示一种药物(靶标)与该类实体的关系。如在药物−疾病关系网络,每种药物由疾病编码,表示为一个二进制向量,向量的每一个字节表示一种疾病,1表示该药物可以治疗该疾病,0则表示不可以或目前还不确定。然后用相似度或距离函数(如Jaccard相似系数、欧氏距离、余弦相似性、GIP(gaussian interaction profile)核函数)衡量两种药物(靶标)之间的相似度。使用不同相似性度量函数或从不同药物(靶标)相关的网络计算得到的药物−药物(靶标−靶标)相似性矩阵可以通过线性组合策略、加权求和、几何平均、算数平均、最大值、非线性融合[190]、相似性网络融合(similarity network fusion,SNF)[191]等方式进行融合。文献[190]详细总结了各种相似性衡量方法和相似性矩阵融合方法。
基于相似性方法的优势在于:1)不必进行复杂的特征提取和特征选择,2)计算化学结构相似性以及基因组序列相似性的度量函数发展较为完善,3)可以直接与核方法相结合,4)相似性度量揭示了药物和基因的联系。该类方法的缺陷在于已知的药物以及药物−靶标相互作用关系数量远远少于未知的数量,无法为一个不存在已知相似关系的新药物(新标靶)预测标靶(药物)。另外,模型的结果和表现会依赖于相似性度量的选取,这是因为相似性定义了药物在高维空间中的相对距离,使用集成学习方法、融合多种度量算法是解决此缺陷的主要思路[15]。
2.3.2 基于随机游走的方法和基于知识图谱的方法
基于随机游走的方法使用DeepWalk[192]、word2vec[65]、node2vec[193]、metapath2vec[194]等算法从异质图中学习药物节点和靶标节点的低维特征表示用于药物−靶标相互作用预测任务[84,189,195]。基于知识图谱的方法将生物医学异质图转换为三元组描述两两生物医学实体之间的关系,然后使用知识图谱嵌入模型(如TransE[196]、DistMult[197]、ComplEx[198])学习实体的特征表示[97,199-201]。
基于随机游走的方法和基于知识图谱的缺陷在于它们学习到的都是浅层的特征表示,也无法利用药物和靶标的结构信息。基于知识图谱的方法不能很好地建模生物医学实体的组合关系,如药物−靶标−疾病−靶标。随着生物医学实体数量的增加,基于随机游走的方法生成的序列数量呈指数增加,增加计算复杂度和计算内存。另外,随着时间推移,不断有新的知识产生。现有的知识图谱嵌入方法由于其优化目标与知识图谱中的所有事实三元组相关,因此每次知识图谱发生变化时都需要重新学习模型。同样的,当有新的实体和关系加入时,基于随机游走的方法也需要重新生成序列和训练模型。
2.3.3 基于深度学习的方法
基于深度学习的方法主要采用GNN模型建模生物实体之间的关系。可以直接将GNN模型(如GAT、GCN)分别作用于每个药物(靶标)相关的网络,然后再聚合从不同网络学习到的药物(靶标)节点特征表示[98-99,157];也可以根据药物−药物相似性、靶标−靶标相似性和已知的药物−靶标相互作用关系构建药物−靶标异质网络,然后再用GNN模型从不同边类型的网络中学习药物和靶标的特征表示并聚合[187,202]。传统GNN模型(如GAT、GCN)的感受野非常小,因为每一层只能聚合一阶邻域节点的特征,而且大多数的GNN模型通常只有两三层,堆叠过多的层数会增加模型的过平滑风险,这就意味着远距离的节点无法有效地传递信息。为了挖掘局部结构和语义信息,一些研究工作利用随机游走策略[97,100,160,165,203]或元路径方法[100,142,152,154,161,174]提取生物医学异质图中的高阶信息。其中,元路径的起始节点和终端节点类型一致,都是药物节点或靶标节点。由随机游走或元路径获得的序列/子图可以采用RNN模型[165]、Transformer[154]或者GNN模型[100,142,152,160-161,174,203]编码节点特征。另外,不同类型的元路径对节点的重要性不一样,常采用注意力机制聚合从不同元路径学习的节点特征[100,152,161,174]。自定义元路径的方式需要专业背景知识,且缺乏灵活性和可扩展性。HampDTI[163]使用注意力机制自动学习元路径,HGAN[203]利用注意力扩散机制学习生物医学异质图中的局部结构信息。此外,为了提高模型的泛化性和提取的特征的判别性,一些研究工作将GNN模型与自监督学习策略相结合[98,142,154,174]。提取的药物特征表示和靶标特征表示可以采用前面介绍的特征融合方式进行融合得到药物−靶标样本特征表示,如拼接[138,165,189]、内积[80,98-99]、将药物−靶标对作为节点,用GNN模型学习节点特征表示[161,174,204]等。
总的来说,基于异质数据的药物−靶标相互作用预测方法主要用于药物重定位。该类方法极大地依赖于数据的完整性,而疾病、副作用等信息要经过长时间的积累才能获得,某些数据特征的缺失会直接限制了这些方法的应用和预测结果。药物重定位面临以下法律和科学难题:1)药物重定位具有机缘巧合性。如癌症、痴呆等疾病,它们的生命机理极其复杂且变幻莫测。随着越来越多的数据可以获得以及人们对药物在生物体内的药理学理解得更充分,一种药物除了可以治疗起初定位的疾病,也可以重新定位治疗其他疾病。许多成功的药物重定位案例都来自偶然发现,如伟哥。因此,预测相关的重定位机遇是一件困难的事情。2)企业和法律层面的问题。药物重定位是以利益驱动的,一种药物的知识产权已经或邻近到期,即使该药物具有较强的药理学特征,重定位研究也不会继续下去。其次,药物重定位并不是标准监管程序的一部分,因此管理问题可能会发生,进而延误或阻止研究进程。另外,药物重定位存在安全争议。重定位的药物对特定人群会有治疗效果,对其他生理状况不同的病人或许会有副作用,因此会大大降低重定位的价值。3)药物剂量问题。重定位后的药物仍然需要满足在低剂量下具有较高的功效。取决于药物在人体结构的目标位置,药物的代谢动力学性质会发生改变。
3. 评价指标
药物−靶标相互作用预测通常建模二分类任务或回归任务。将预测药物−靶标相互作用关系作为二分类任务时,每个样本的真实标签用0/1表示,样本的预测结果根据实际情况分为4类,分别是真正例(true positive,TP)(预测值为1,真实值为1),假正例(false positive,FP)(预测值为1,真实值为0),真反例(true negative,TN)(预测值为0,真实值为0)和假反例(false negative,FN)(预测值为0,真实值为1)。常用的模型评价指标包括真阳性率(true positive rate,TPR)、伪阳性率(false positive rate,FPR)、准确率(accuracy,AC)、精确率(precision,PR)、召回率(recall,RC)、F1、灵敏度(sensitivity,SS)、特异度(specificity,SP)、MCC(Matthews correlation coefficient)、ROC(receiver operating characteristic)曲线、AUC(area under the ROC curve)、AUPR(area under the precision-recall curve)等。
将预测药物−靶标相互作用作为回归任务时,样本
$ i $ 的实际标签$ {y}_{i} $ 和预测标签$ {\check{y}}_{i} $ 都是连续值,常用的模型评价指标包括均方根误差(root mean squared error,RMSE)、平均绝对误差(mean absolute error,MAE)、一致性指数(concordance index,CI)、均值回归(regression toward the mean)(又称$ {r}_{m}^{2} $ 指数)、R-squared($ {R}^{2} $ ,又称可决系数(coefficient of determination))、皮尔逊相关系数(Pearson correlation coefficient,PCC)、线性回归中的标准偏差(standard deviation (SD) in linear regression)等。表1总结了药物−靶标相互作用预测任务常用到的评价指标。表 1 药物−靶标相互作用预测任务用到的评价指标Table 1 Metrics used in drug-target interaction prediction序号 评价指标 表达式 描述 1 TPR TP / (TP + FN) 正类数据被分为正类的比例 2 FPR FP / (FP + TN) 负类数据被分为正类的比例 3 AC (TP + TN) / (TP + FN + FP + TN) 对于样本不平衡数据集,准确率会失效 4 PR TP / (TP + FP) 查准率,正类数据在预测为正类中的比例 5 RC/SS TP / (TP + FN) 查全率,同TPR 6 SP TN / (FP + TN) 描述识别出的负例占所有负例的比例 7 MCC $\dfrac{\left(\left(\mathrm{T}\mathrm{P}\times \mathrm{T}\mathrm{N}\right)-\left(\mathrm{F}\mathrm{P}\times \mathrm{F}\mathrm{N}\right)\right)}{\sqrt{\left(\mathrm{T}\mathrm{P}+\mathrm{F}\mathrm{P}\right)\left(\mathrm{T}\mathrm{P}+\mathrm{F}\mathrm{N}\right)\left(\mathrm{T}\mathrm{N}+\mathrm{F}\mathrm{P}\right)\left(\mathrm{T}\mathrm{N}+\mathrm{F}\mathrm{N}\right)} }$ 衡量预测结果质量,值介于−1∼1 8 F1 2 × (PR × RC) / (PR + RC) 精确率和召回率的调和平均值 9 ROC 以TPR为纵坐标,FPR为横坐标绘制的曲线 10 AUC ROC曲线下的面积,表示预测的正例排在负例前面的概率 11 AUPR 以PR为纵坐标,RC为横坐标的曲线下的面积 12 RMSE $\sqrt{\dfrac{1}{N}\displaystyle\sum _{i=1}^{N}{\left({y}_{i}-{\check{y} }_{i}\right)}^{2} }$ 量化回归模型整体误差,值越小,模型拟合效果越好 13 MAE $\dfrac{1}{N}\displaystyle\sum _{i=1}^{N}\left|{y}_{i}-{\check{y} }_{i}\right|$ 衡量预测值与真实值的距离,值越小,模型拟合效果越好 14 CI $\dfrac{1}{Z}\displaystyle\sum _{i,j,i > j}\sigma ({y}_{i} > {y}_{j})\varphi ({\check{y} }_{i}-{\check{y} }_{j})$
$\sigma \left(x\right)=\left\{ \begin{array}{c}\begin{array}{cc}1& x\ge 0\end{array}\\ \begin{array}{cc}0& x < 0\end{array}\end{array}\right.$
$ \varphi \left(x\right)=\left\{ \begin{array}{c}\begin{array}{cc}1& x > 0\end{array}\\ \begin{array}{cc}0.5& x=0\end{array}\\ \begin{array}{cc}0& x < 0\end{array}\end{array}\right. $衡量预测结果与实际结果相一致的概率,值介于0∼1 15 $ {R}^{2} $ $1-\dfrac{ \displaystyle\sum _{i=1}^{N}{\left({y}_{i}-{\check{y} }_{i}\right)}^{2} }{ \displaystyle\sum _{i=1}^{N}{\left({y}_{i}-\overline{y}\right)}^{2} }$ 衡量真实值与拟合回归线之间的接近程度,值介于0∼1 16 PCC $\dfrac{ \displaystyle\sum _{i=1}^{N}\left({y}_{i}-\overline{y}\right)\left({\check{y} }_{i}-\overline{\check{y} }\right)}{ \displaystyle\sum _{i=1}^{N}{\left({y}_{i}-\overline{y}\right)}^{2}{\left({y}_{i}-\overline{\check{y} }\right)}^{2} }$ 度量预测值与真实值的线性相关程度,值介于−1∼1 17 $ {r}_{m}^{2} $ $ {r}^{2}\times (1-\sqrt{{r}^{2}-{r}_{0}^{2}}) $
$ r $= PCC
${r}_{0}=1-\dfrac{ \displaystyle\sum _{i=1}^{N}{({y}_{i}-k*{\check{y} }_{i})}^{2} }{ \displaystyle\sum _{i=1}^{N}{({y}_{i}-\overline{y})}^{2} }$反映模型的预测潜力 18 SD $\sqrt{\dfrac{1}{N-1}\displaystyle\sum _{i=1}^{N}{\left[{y}_{i}-\left(a+b{\check{y} }_{i}\right)\right]}^{2} }$ 量化回归模型拟合误差 4. 数据来源
目前,有许多药物相关的数据库/数据集已建立,可以支撑药物−靶标相互作用预测任务。本节介绍在药物−靶标相互作用预测任务中常用到的数据库和数据集。为了便于区分,在本文中数据库指开源的、会定期更新数据的网站,预测任务需要的药物−靶标相互作用关系以及药物相关的数据和靶标相关的数据可以从这些网站获取。而数据集包含药物−靶标相互作用关系,可直接用于预测任务或预处理后可用于预测任务。根据数据库的主要聚焦范围,本文将相关的数据库划分为4类,即药物−靶标相互作用数据库、药物相关的数据库、靶标相关的数据库和支撑数据库。统计截止时间为2023年8月4日。
4.1 药物−靶标相互作用数据库
药物−靶标相互作用数据库收集药物−靶标相互作用信息以及其他相关的信息。文本列出11个可用于药物−靶标相互作用预测任务的数据库,包括ChEMBL、DrugBank、STITCH、BindingDB、PDBbind、LINCS、GtoPdb、IntAct、DGIdb、Promiscuous-2.0和DTP。
ChEMBL[205](https://www.ebi.ac.uk/chembl/)是具有类药物特性生物活性分子的数据库,由欧洲分子生物学试验所−欧洲生物医学研究学(European Molecular Biology Laboratory-European Bioinformatics Institute,EMBL-EBI)维护。ChEMBL汇集了化学、生物活性和基因组数据以帮助将基因组信息转化为有效的新药。最新版本ChEMBL-33在2023年5月更新,约包括240万个化合物、1.4万个药物、1.5万个靶标。除此之外,ChEMBL还提供药物作用机制、适用症、分析(assay)、细胞、组织等信息。
DrugBank[206](https://go.drugbank.com/)是一个生物信息学和化学信息学数据库,在2006年由加拿大阿尔伯塔大学成立,是使用最为广泛的数据库之一。DrugBank包含详细的药物信息(如药物分子信息、药理学信息)、药物靶标信息(如序列、结构、路径)、药物−靶标相互作用关系、药物−药物作用关系和药物−食物作用关系等信息,其中,药物包括FDA批准通过的药物和进入FDA批准流程的实验药物。DrugBank-5.0增加了大量新的数据,包括药物代谢组学、药物转录组学和药物蛋白组学信息。最新发布的DrugBank-5.1.10(2023-01-04)包含15 685个药物(2 744个批准的小分子药物、1 586个生物制剂、124个保健药物、超过6 720个实验药物)和5 295个蛋白质(药物靶标、酶、转运体、载体)。
STITCH(Search Tool for Interactions of Chemicals)[207](http://stitch.embl.de)是一个存储化学分子和蛋白相互作用关系的数据库,最近版本STITCH-5发布于2016年。STITCH收集的相互作用关系包括直接(物理)和间接(功能)关系,主要来源于5个方面:基因组上下文预测、高通量实验、(保守)共表达、文本挖掘、数据库中的先前知识。除了内部预测和同源性转以外,STITCH还依赖于其他数据库资源,如PubChem、KEGG、PDB、DrugBnak等。
BindingDB[208](https://www.bindingdb.org/)提供药物−靶标结合亲和力数据,所有这些数据都来自科技文献和专利。BindingDB在2000年首次发布,最近的更新时间是2023年7月31日,包括120万个化合物和9 200个靶标,共有280万个结合亲和力数据。
PDBbind[209]为来自PDB(Protein Data Bank)数据库的生物分子复合体提供实验结合亲和力数据,包括
$ {K}_{i} $ 、$ {K}_{d} $ 和$ {\mathrm{I}\mathrm{C}}_{50} $ 。生物分子复合体包括蛋白−配体、蛋白−蛋白、蛋白−核酸以及核酸−配体。PDBbind首次发布在2004年5月,最新版本PDBbind-2020在2020年8月23日发布,包括19 443个蛋白−配体复合体样本。PDBbind中有效的复合体划分为3个重叠的数据集,“general set”“refined set” 和“core set”“refined set”是从“general set”挑选的高质量蛋白−配体复合体,通常作为训练集训练模型。“core set”是CASF(comparative assessment of scoring functions)的基准数据集,通常作为测试集。LINCS(Integrated Network-Based Cellular Signatures)[210](https://lincsproject.org/)旨在通过分类基因表达变化和其他细胞暴露于各种干扰时发生的细胞过程变化建立一个基于网络的生物学理解。LINCS一种收集了424个数据集,其中180个数据集是KINOMEscan激酶−小分子结合分析。
GtoPdb(https://blog.guidetopharmacology.org/)[211]最初由英国药理学学会(British Pharmacologic-al Society,BPS)和国际基础和临床药理学联盟(International Union of Basic and Clinical Pharmacology,IUPHAR)联合开发,现在与威康信托基金共同开发,旨在提供所有药理学靶标的简明概述。GtoPdb-2023.1(2023年4月26日发布)包含3 023个人类靶标(1 662个与配体有相互作用)、11 944个配体(8 814个与靶标有相互作用)、19 890个配体−靶标结合常数。GtoPdb还包括指向其他资源的链接,例如Ensembl、UniProt、PubChem、ChEMBL、DrugBank等。
IntAct[212](https://www.ebi.ac.uk/intact/)是一个提供分子相互作用关系的免费开源数据库。IntAct共包括16个分子相互作用数据集,包括亲和组学、蛋白与疾病(如阿尔茨海默病、癌症、冠状病毒、炎性肠病、糖尿病等)的关系、蛋白−蛋白相互作用关系。IntAct中所有的相互作用关系均来自文献挖掘或用户的提交。
DGIdb(Drug-Gene Interaction Database)[213](https://www.dgidb.org/)提供药物−基因相互作用关系以及可用药基因组信息,这些信息来自发表物、其他数据库和网络资源,共计41个资源。DGIdb在2013年首次发布,最新版本在2022年2月发布,包含4万多个基因、1万个药物、10万多个药物−基因相互作用关系。
Promiscuous-2.0[214](https://bioinf-applied.charite.de/promiscuous2/)是用于药物重定位的一站式资源,包括药物(约100万个)、靶标(约1万个)、副作用(约11万个)和适用症4种类型实体以及实体之间的关系,其中,药物−靶标相互作用关系约有300万个。此外,Promiscuous-2.0还提供了一个网络表示总结实体之间的关系。
DTP[215](drug target profiler)(http://drugtargetprofiler.fimm.fi)是一个探索药物−靶标相互作用的交互式网络应用程序,用于指导药物重定位和药物作用方式研究。DTP包括化合物近94万个,靶标蛋白5 077个,药物−靶标相互作用关系约443万个,其中,药物靶标相互作用基于剂量反应测量,例如
$ {K}_{i} $ 、$ {K}_{d} $ 、$ {\mathrm{I}\mathrm{C}}_{50} $ 等。4.2 以药物为核心的数据库
本文介绍5个以药物为核心的数据库,包括PubChem、DrugComb、DrugCombDB、GDSC、DrugCentral。
PubChem[216](https://pubchem.ncbi.nlm.nih.gov/)从近千个数据资源收集分子信息,包括化学结构、标识符、化学和物理特性、生物活性、安全、毒性等。PubChem中大部分是小分子,也包括大分子,如核苷酸、脂类、多肽类、碳水化合物和化学修饰的生物聚合物。PubChem包括化合物约1.16亿个,蛋白质约19万个,生物活性约3亿个。
DrugComb[217](https://drugcomb.org/)是一个药物组合筛选分析网站,收集、标准化和协调各种癌症和其他疾病(如疟疾、COVID-19)的药物组合筛选研究结果。DrugComb也提供网络建模工具用于可视化给定癌症样本的药物作用机制和药物组合。DrugComb1.5(2021年更新)包括8 397个药物、2 320个细胞系、33个组织、约74万个药物组合。
DrugCombDB[218](http://drugcombdb.denglab.org/)也是一个药物组合数据库,药物组合数据主要来源于3个方面:药物组合高通量筛选实验、外部数据库、PubMed文献挖掘。DrugCombDB最近的更新时间在2019年,包括448 555种药物组合,涵盖2 887个药物,124个细胞系。
GDSC(Genomics of Drug Sensitivity in Cancer Project)[219](https://www.cancerrxgene.org/)由威康桑格研究所(英国)的癌症基因组项目和麻省总医院癌症中心(美国)的分子治疗中心合作创建,收集癌细胞药物敏感性和药物反应分子标记物信息。最近版本GDSC-8.4在2022年7月发布,鉴定了1 000种癌细胞特征,并用100种化合物进行了筛选。
DrugCentral[220](https://drugcentral.org/)提供活性成分化学实体、药品、适应症药物作用方式、药理作用等信息。DrugCentral定期监控FDA、EMA和PMDA以获取新批准的药物。2022年9月9日更新的DrugCentral包括4 927个活性成分,其中4 080个是小分子,374个是生物制剂。
4.3 以靶标为核心的数据库
本文介绍9个以靶标为核心的数据库,包括Ensembl、UniProt、KEGG、BioGRID、CCLE、CellMinerCDB、PDB、STRING、HPRD。
Ensembl[221](https://www.ensembl.org/)提供关于脊椎动物的高质量基因组数据。Ensembl产生高质量的基因组注释,包括基因、变异、调控区域和比较基因组学资源。此外,Ensembl还提供了13种数据处理工具,如VEP(variant effect predictor)(分析变异,预测已知和未知变异的功能结果)、BLAST/BLAT(为用户提供的DNA或蛋白序列搜索基因组)、BioMart(是一种数据挖掘工具,从Ensemble中导出自定义数据集)等。最新版本Ensembl-110在2023年7月份发布。
UniProt(Universal Protein Resource)[222](https://www.uniprot.org/)是一个全面的高质量蛋白序列与功能信息数据库,包括4个核心数据库,即UniProtKB(UniProt Knowledgebase)、Proteomes、UniRef(UniProt Reference Clusters)和UniParc(UniProt Archive)。Proteomes包含所有具有序列基因组物种的蛋白质集合,UniRef按照100%、90%和50%一致性对蛋白序列聚类,UniParc记录蛋白序列的数据来源。UniProtKB提供蛋白序列与功能信息,包括验证过的蛋白集合(Swiss-Prot,约57万个)和未验证过的蛋白集合(TrEMBL,约2.5亿个)两部分。最近版本UniProt-2023-03在2023年6月28日发布。
KEGG(kyoto encyclopedia of genes and genomes)[223](https://www.kegg.jp/kegg/)是一个集成数据库资源,在1995年由日本人类基因组计划首次提出,用于从基因组和分子水平信息中了解生物系统的高级功能和效用,如细胞、生物体和生态系统。最新版本KEGG-107.0在2023年7月1日更新。KEGG大致可以划分为4大类,每类包括若干个数据库,即系统信息(PATHWAY、BRITE、MODULE)、基因组信息(ORTHOLOGY (KO)、GENES、GENOME)、化学信息(COMPOUND、GLYCAN,REACTION、RCLASS、ENZYME)和健康信息(NETWORK、VARIANT、DISEASE、DRUG、DGROUP)。DRUG数据库包含日本、美国和欧洲批准通过的药物的信息,如靶标、代谢及其他分子相互作用网络信息。
BioGRID[224](biological general repository for interaction datasets)(https://thebiogrid.org/)存储多种物种的蛋白质和基因相互作用关系,包括人类、老鼠、酵母、蠕虫和苍蝇。BioGRID中所有数据均来自生物医学文献报告的实验结果,包括低通量研究和高通量数据集。此外,BioGRID也包括蛋白转录后修饰以及蛋白或基因与生物活性小分子的相互作用关系数据。2023年7月1日发布BioGRID-4.4.223从大约8.3万篇文献中挖掘出约264万个蛋白质和基因相互作用关系,3万多个分子相互作用关系,约113万个模式生物物种的转录后修饰。
CCLE(cancer cell line encyclopedia)[225](https://sites.broadinstitute.org/ccle/)致力于从来自同组织群系的近1 000个细胞系中生成大规模分析数据集。CCLE通过质谱法(mass spectrometry)从375个细胞系中收集数据,包括基因表达、DNA拷贝数、混合捕捉测序、组蛋白分析、RNA-seq、DNA甲基化、miRNA分析、全基因组测序、代谢物分析等信息。
CellMinerCDB(CellMiner Cross-Database)[226](https://discover.nci.nih.gov/rsconnect/cellminercdb/)是一个交互式网络应用程序,整合不同来源的癌细胞系药物基因组数据。2023年7月发布的CellMinerCDB-1.8包含多种类型的数据,如药物活性、mRNA表达、RNA-seq表达、DNA甲基化、DNA变异、DNA拷贝数、MicroRNA、蛋白等。
PDB(protein data bank)[227](https://www.wwpdb.org/)始建于1971年,专门收录蛋白质、DNA、RNA以及它们与金属离子、药物和其他小分子结合复合体的三维结构信息。自2003年起,PDB由wwPDB(worldwide protein data bank)、RCSB PDB(US research collaboratory for structural bioinformatics protein data bank)(https://www.rcsb.org/)、PDBe(protein data bank in Europe)(https://www.ebi.ac.uk/pdbe/)、PDBj(protein data bank Japan)(https://pdbj.org/)和BMRB(BioMagResBank)(https://www.ebi.ac.uk/emdb/)共同维护。
STRING[228](https://string-db.org/)系统地收集和整合蛋白−蛋白的物理关系和功能关系。数据来源包括科技文献文本挖掘、高通量实验、从共表达中得到的计算交互作用预测、保守基因组背景、数据库中的已有知识。当前版本STRING-12大约包括6 800万个蛋白质。
HPRD(human protein reference database)[229](http://www.hprd.org/)是一个收集人类蛋白质组学信息的数据库,所有信息均由生物医学专家从公开文献中提取。HPRD最后一次更新时间是在2009年1月16日。
4.4 支撑数据库
Open Targets[230](https://www.opentargets.org/)利用人类遗传学和基因组学数据进行系统的药物靶点识别和优化。Open Targets平台(https://platform.opentargets.org/)主要关注靶标−疾病关系,通过构建和打分靶标−疾病关系来帮助药物靶标识别和优化。同时,Open Targets平台也提供靶标、疾病、表型、药物以及这些实体之间的关系数据。Open Targets平台每年更新5次,最近版本23.06在2023年6月26日发布。
DisGeNET[231](https://www.disgenet.org/)收集与人类疾病相关的基因和变异信息,数据来源包括GWAS目录、科技文献等。DisGeNET涵盖了人类所有疾病以及正常和异常特征,核心数据是基因−疾病关系和变异−疾病关系。目前的版本DisGeNET-7.0大约包括基因-疾病关系114万个(由约2.2万个基因和3万个疾病组成),变异−疾病关系37万个(由约20万个变异和1.4万个疾病组成)。
COSMIC(catalogue of somatic mutations in cancer)[232](https://cancer.sanger.ac.uk/cosmic)收集与人类癌症相关的体细胞突变信息数据,主要包括由专家手工提取的高精度数据和全基因组筛选数据两大类。最新版本COSMIC-98更新于2023年5月23日。
SIDER(side effect resource)[233](http://sideeffects.embl.de/)从公开文献中收集上市药物及其相关的副作用信息,包括副作用频率、药物和副作用分类以及药物−靶标相互作用关系。当前版本SIDER-4.1(2015年10月21日发布)包括1 430个药物,5 868个副作用,139 756个药物−副作用关系。
CTD(comparative toxicogenomics database)[234](http://ctdbase.org/)的目的是加强人们对环境暴露对人类健康影响的了解,识别环境−疾病联系。CTD收集化学、疾病、基因、表型以及化学分子−基因/蛋白相互作用、化学分子−疾病关系、化学分子−表型相互作用、基因−疾病关系、基因−基因相互作用等数据,并将这些数据与功能和路径数据相结合来帮助制定有关受环境影响的疾病的潜在机制的假说。CTD在2004年11月12日首次发布,最近版本CTD-17142在2023年7月31日发布。
4.5 数据集
Davis数据集[235]、KIBA(kinase inhibitor bioactivity)数据集[236]和Metz数据集[237]聚焦激活酶(kinase)蛋白家族的生物活性,是预测药物−靶标结合亲和力的基准数据集。激活酶在许多癌症和炎症性疾病的细胞信号转导过程中起到至关重要的作用。Davis数据集包括72个药物分子和442个靶标,共组成31 824个药物−靶标相互作用对。药物−靶标对的亲和力指标是
$ {K}_{d} $ ,$ {K}_{d} $ 值在研究工作中通常转换到对数空间($ {pK}_{d}=-{\mathrm{log}}_{10}({K}_{d}/{10}^{9}) $ 。KIBA数据集最初包含52 498个药物分子,467个靶标,246 088个药物−靶标相互作用关系。He等[78]去除具有相互作用关系不足10个的药物分子和靶标,得到2 116个药物分子和229个靶标,目前大部分研究工作采用过滤后的数据集。KIBA数据集中的激活酶抑制生物活性包括$ {\mathrm{I}\mathrm{C}}_{50} $ 、$ {K}_{i} $ 和$ {K}_{d} $ ,在实际研究工作中为优化不同指标间的一致性,通常将这些亲和力值转换为KIBA得分并转换到对数空间[236]。Metz数据集包括1 421个药物分子和156个靶标,42%的药物−靶标对的亲和力是$ {pK}_{i} $ ($ {K}_{i} $ 的对数值)。Yamanishi08数据集[238]在2008年公布,是预测药物−靶标相互作用关系的基准数据集,药物−靶标相互作用关系来自KEGG BRITE、BRENDA、SuperTarget和Drugbank数据库。根据靶标蛋白的类型,药物−靶标相互作用关系划分E(enzymes)、IC(ion channels)、GPCR(G-protein-coupled receptors)和NR(nuclear receptor)4个子集,分别包括2 926个(445个药物,664个靶标)、1 476个(210个药物,204个靶标)、635个(223个药物,95个靶标)、90个(54个药物,26个靶标)药物−靶标相互作用关系。Chu等[43]在Yamanishi08数据集的基础上,从KEGG BRITE、UniProt和DrugBank数据库收集了新的药物、靶标以及药物−靶标相互作用数据,更新后的四个子集分别包括7 371个(1 777个药物,1 411个靶标)、6 385个(765个药物,238个靶标)、5 383个(1 680个药物,156个靶标)和886个(541个药物,33个靶标)药物−靶标相互作用关系。
Luo等[158]公布了一个异质数据集用于药物−靶标相互作用预测研究。该数据集从DrugBank(version 3.0)、HPRD(release 9)、CTD(2013)和SIDER(version 2)数据库收集而来,包括4种生物医学实体(药物、靶标、疾病和副作用)和6种实体之间的关系(药物−靶标、药物−药物、药物−疾病、药物−副作用、蛋白−疾病以及蛋白−蛋白相互作用关系),共有12 015个实体,1 895 445个实体相互作用关系。在过去的十多年里,大量的药物−靶标相互作用关系以及其他实体关系被发现,但该异质数据集并没有包含这些信息。正样本数据的缺失不仅会在数据建模过程中引入误差,也会使模型评估具有较高假阴率的潜在风险。针对此问题,Li等[161]从DrugBank(version 5.1.8)、UniProtPK(release 20214)、CTD(2021)、SIDER(version4)和STRING(version 11.5)数据库整理了一个异质数据集,同样包括4种生物医学实体和6种实体之间的关系,共有15 322个实体,5 126 875个实体相互作用关系。Huang等[239]构建了TDC(therapeutics data commons)系统,该系统包括66个数据集,涉及22个任务,可直接用于药物研发相关任务的研究工作。TDC系统提供了TDC.BindingDB、TDC.DAVIS和TDC.KIBA等3个数据集用于药物−靶标结合亲和力预测任务,并提供了多种数据划分方式(药物冷启动、靶标冷启动)和模型验证指标。
5. 研究挑战与展望
过去十年,得益于大数据、人工智能、计算技术的快速发展,药物−靶标相互作用预测的准确性和效率都得到前所未有的提高。尽管如此,药物−靶标相互作用预测仍面临以下挑战。
1)现有数据库只包含具有相互作用关系的药物−靶标对(正样本)而未提供不具有相互作用关系的样本信息(负样本)。许多监督训练模型简单的将未标记相互作用关系的药物−靶标对作为负样本,降低了模型的预测准确性。而且负样本的数量远远大于正样本的数量,正负样本不均衡问题也是深度学习模型面临的一个挑战。在实际场景中,对于一个蛋白,大多数的药物分子都是负样本。因此,随机过采样算法常用来增加训练集中正样本的比例,但这也会影响模型在实际应用的泛化能力。研究半监督/自监督学习模型/算法,增加数据库中的负样本数据可有效解决这一问题。此外,现有的基准数据集在划分活性和非活性分子可能存在偏差,而深度学习模型很容易学习到这种偏差。决定生物活性的实验方法和标准各种各样,一些活性分子也缺乏可做定量比较的定量活性数据。从不同实验阶段和设备获得的异质生物医学数据带有噪声,因此需要对发现、获取、整合和重利用药物−靶标相互作用数据提供指导方针,建立包含活性和非活性分子的高质量无偏基准数据集。
2)蛋白质的三维折叠结构决定蛋白的功能,基于三维结构的药物−靶标相互作用预测模型为药物和靶标的物理相互作用提供了较为直接的表征。但是目前的药物−靶标相互作用预测模型多关注药物和蛋白质的一维和二维数据,忽略了三维空间的结构信息。另外,基于结构的药物−靶标相互作用预测方法面临一些蛋白质没有三维结构信息的问题。同源建模或一些蛋白结构预测软件获得的蛋白质三维结构活能会带来偏差。AlphaFold II在蛋白质结构预测取得了重大突破。未来,蛋白质的三维结构将会更易于获得,基于结构的药物−靶标相互作用预测模型也会取得更加准确的预测结果。
3)生物医学交互网络分为二部图和异质图。二部图的优点是简单,节点只有药物和靶标,不需要结合多种交互作用信息。由于许多药物和靶标的相互作用关系还未得到验证,存在信息缺失问题,因此基于药物−靶标二部图方法的预测结果有限。一些研究工作将其他药物有关的信息和靶标有关的信息整合到二部图中形成异质图,在一定程度上填补了链路缺失的空白。与二部图相比,异质图结合了生物医学实体不同类型的交互信息,为预测药物−靶标相互作用关系提供了多视角信息。但是,异质图建模面临两个挑战。一方面异质图具有较高的建模理论要求,另一方面,网络结构的优化需要结合不同来源的数据,例如疾病、副作用信息。未来,基于异质数据的药物−靶标相互作用预测研究有待进一步探索。
4)深度学习模型就像一个黑盒子,缺乏可解释性,而可解释性在生物医学领域非常有必要。具有可解释性的模型不仅可以预测药物−靶标相互作用,也将可以帮助人们更好地理解模型潜在作用机制,帮助发现新的活性分子和新的靶标。因此,未来可以从因果推理、可解释性角度为药物−靶标相互作用预测任务提出解决方案。
5)实际上,所有的蛋白都是高度动态生物分子,不断地与其周围产生相互作用,并生成各种各样的构象状态。因此,蛋白存在一系列集成构象状态。但是,实验解析出的结构仅仅是最常见构象状态的临时平均构象状态,因此无可避免地会丢失结构的动态效应。此外,配体部分溶于溶剂,因此配体不仅与蛋白产生相互作用,也与溶剂(例如水,缓冲离子)发生相互作用。在分子动力学模拟时长足够的情况下,配体−溶剂相互作用以及其他的长/短范围相互作用对配体构象的影响便可以自然地嵌入到分子动力学轨迹中[240]。
6)当前的深度学习技术更适合在确定性环境下解决单领域、单任务问题,药物研发仍存在不确定性,知识不够完备,而且需要创造性和灵活性。从人工智能的角度来讲,当前,该领域处于第三次浪潮的初始阶段,这一波人工智能的发展主要受到大数据,机器学习尤其是深度学习技术的推动,整体上还处于弱人工智能阶段,此阶段的人工智能仍然有一些局限性。应用场景必须满足具有丰富的数据或知识、完备信息、确定性信息、静态(或按确定性规律演化)、单领域和单任务5个条件。总体来讲,当前深度学习技术解决此类问题时所需条件严苛,局限性大,缺乏灵活性。
6. 结束语
药物−靶标相互作用预测是药物筛选和药物重定位的关键环节。研究基于深度学习的药物−靶标相互作用预测不仅具有学术研究,同时也具有现实应用意义和商业价值。得益于深度学习技术的快速发展,药物−靶标相互作用预测技术复杂多样,为研究者快速了研究现状并从中选用合适的方法带来挑战。本文从使用的数据的种类、问题定义、数据表示、特征学习与特征融合、预测模块、学习方式、实验设置等多角度多层次分析现有药物−标靶相互作用预测方法,从而帮助药物研发领域研究者全面了解深度学习在药物−靶标相互作用预测领域的最新研究进展,进而提高研究效率和研究质量。
-
图 1 药物−靶标相互作用类比示意
Fig. 1 Illustration of drug-target interaction
下载:
全尺寸图片
图 2 药物−靶标相互作用过程
Fig. 2 Process of drug-target interaction
下载:
全尺寸图片
图 3 药物重定位依据
Fig. 3 Opportunities of drug repositioning
下载:
全尺寸图片
图 4 药物和靶标相互作用预测方法总结
Fig. 4 Summary of drug-target interaction prediction
下载:
全尺寸图片
图 5 药物数据表示
Fig. 5 Representations of drugs
下载:
全尺寸图片
图 6 靶标数据表示
Fig. 6 Representations of targets
下载:
全尺寸图片
图 7 药物−靶标相互作用预测流程
Fig. 7 Flowchart of drug-target interaction prediction
下载:
全尺寸图片
图 8 不同生物医学概念关系
Fig. 8 Relationships of different biomedical concepts
下载:
全尺寸图片
表 1 药物−靶标相互作用预测任务用到的评价指标
Table 1 Metrics used in drug-target interaction prediction
序号 评价指标 表达式 描述 1 TPR TP / (TP + FN) 正类数据被分为正类的比例 2 FPR FP / (FP + TN) 负类数据被分为正类的比例 3 AC (TP + TN) / (TP + FN + FP + TN) 对于样本不平衡数据集,准确率会失效 4 PR TP / (TP + FP) 查准率,正类数据在预测为正类中的比例 5 RC/SS TP / (TP + FN) 查全率,同TPR 6 SP TN / (FP + TN) 描述识别出的负例占所有负例的比例 7 MCC $\dfrac{\left(\left(\mathrm{T}\mathrm{P}\times \mathrm{T}\mathrm{N}\right)-\left(\mathrm{F}\mathrm{P}\times \mathrm{F}\mathrm{N}\right)\right)}{\sqrt{\left(\mathrm{T}\mathrm{P}+\mathrm{F}\mathrm{P}\right)\left(\mathrm{T}\mathrm{P}+\mathrm{F}\mathrm{N}\right)\left(\mathrm{T}\mathrm{N}+\mathrm{F}\mathrm{P}\right)\left(\mathrm{T}\mathrm{N}+\mathrm{F}\mathrm{N}\right)} }$ 衡量预测结果质量,值介于−1∼1 8 F1 2 × (PR × RC) / (PR + RC) 精确率和召回率的调和平均值 9 ROC 以TPR为纵坐标,FPR为横坐标绘制的曲线 10 AUC ROC曲线下的面积,表示预测的正例排在负例前面的概率 11 AUPR 以PR为纵坐标,RC为横坐标的曲线下的面积 12 RMSE $\sqrt{\dfrac{1}{N}\displaystyle\sum _{i=1}^{N}{\left({y}_{i}-{\check{y} }_{i}\right)}^{2} }$ 量化回归模型整体误差,值越小,模型拟合效果越好 13 MAE $\dfrac{1}{N}\displaystyle\sum _{i=1}^{N}\left|{y}_{i}-{\check{y} }_{i}\right|$ 衡量预测值与真实值的距离,值越小,模型拟合效果越好 14 CI $\dfrac{1}{Z}\displaystyle\sum _{i,j,i > j}\sigma ({y}_{i} > {y}_{j})\varphi ({\check{y} }_{i}-{\check{y} }_{j})$
$\sigma \left(x\right)=\left\{ \begin{array}{c}\begin{array}{cc}1& x\ge 0\end{array}\\ \begin{array}{cc}0& x < 0\end{array}\end{array}\right.$
$ \varphi \left(x\right)=\left\{ \begin{array}{c}\begin{array}{cc}1& x > 0\end{array}\\ \begin{array}{cc}0.5& x=0\end{array}\\ \begin{array}{cc}0& x < 0\end{array}\end{array}\right. $衡量预测结果与实际结果相一致的概率,值介于0∼1 15 $ {R}^{2} $ $1-\dfrac{ \displaystyle\sum _{i=1}^{N}{\left({y}_{i}-{\check{y} }_{i}\right)}^{2} }{ \displaystyle\sum _{i=1}^{N}{\left({y}_{i}-\overline{y}\right)}^{2} }$ 衡量真实值与拟合回归线之间的接近程度,值介于0∼1 16 PCC $\dfrac{ \displaystyle\sum _{i=1}^{N}\left({y}_{i}-\overline{y}\right)\left({\check{y} }_{i}-\overline{\check{y} }\right)}{ \displaystyle\sum _{i=1}^{N}{\left({y}_{i}-\overline{y}\right)}^{2}{\left({y}_{i}-\overline{\check{y} }\right)}^{2} }$ 度量预测值与真实值的线性相关程度,值介于−1∼1 17 $ {r}_{m}^{2} $ $ {r}^{2}\times (1-\sqrt{{r}^{2}-{r}_{0}^{2}}) $
$ r $= PCC
${r}_{0}=1-\dfrac{ \displaystyle\sum _{i=1}^{N}{({y}_{i}-k*{\check{y} }_{i})}^{2} }{ \displaystyle\sum _{i=1}^{N}{({y}_{i}-\overline{y})}^{2} }$反映模型的预测潜力 18 SD $\sqrt{\dfrac{1}{N-1}\displaystyle\sum _{i=1}^{N}{\left[{y}_{i}-\left(a+b{\check{y} }_{i}\right)\right]}^{2} }$ 量化回归模型拟合误差 -
[1] BENJAMINE B. 药物研发基本原理[M]. 第四版. 白仁仁, 译. 北京: 科学出版社, 2021. [2] ASHBURN T T, THOR K B. Drug repositioning: identifying and developing new uses for existing drugs[J]. Nature reviews drug discovery, 2004, 3(8): 673–683. doi: 10.1038/nrd1468 [3] ADASME M F, PARISI D, SVESHNIKOVA A, et al. Structure-based drug repositioning: potential and limits[J]. Seminars in cancer biology, 2021, 68: 192–198. doi: 10.1016/j.semcancer.2020.01.010 [4] PARK K. A review of computational drug repurposing[J]. Translational and clinical pharmacology, 2019, 27(2): 59–63. doi: 10.12793/tcp.2019.27.2.59 [5] MOHS R C, GREIG N H. Drug discovery and development: role of basic biological research[J]. Alzheimer’s & dementia, 2017, 3(4): 651–657. [6] KATO S, MOULDER S L, UENO N T, et al. Challenges and perspective of drug repurposing strategies in early phase clinical trials[J]. Oncoscience, 2015, 2(6): 576–580. doi: 10.18632/oncoscience.173 [7] 刘聪. 2021中国AI/计算制药产业报告: 药物发现篇[R]. 北京: 亿欧智库. 2021. LIU Cong. 2021 China AI/computational pharmaceutical industey report: Drug discovery and development[R]. Beijing: EqualOcean Intelligence, 2021. [8] MULLARD A. 2022 FDA approvals[J]. Nature reviews drug discovery, 2023, 22(2): 83–88. doi: 10.1038/d41573-023-00001-3 [9] 翟永杰, 张智柏, 王亚茹. 基于改进TransGAN的零样本图像识别方法[J]. 智能系统学报, 2023, 18(2): 352–359. doi: 10.11992/tis.202111002 ZHAI Yongjie, ZHANG Zhibai, WANG Yaru. An image recognition method of zero-shot learning based on an improved TransGAN[J]. CAAI transactions on intelligent systems, 2023, 18(2): 352–359. doi: 10.11992/tis.202111002 [10] 陈斌, 朱晋宁. 双流增强融合网络微表情识别[J]. 智能系统学报, 2023, 18(2): 360–371. CHEN Bin, ZHU Jinning. Micro-expression recognition based on a dual-stream enhanced fusion network[J]. CAAI transactions on intelligent systems, 2023, 18(2): 360–371. [11] 亢洁, 刘威. 面向装修案例智能匹配的跨模态检索方法[J]. 智能系统学报, 2022, 17(4): 714–720. KANG Jie, LIU Wei. A crossmodal retrieval method for intelligent matching of decoration cases[J]. CAAI transactions on intelligent systems, 2022, 17(4): 714–720. [12] LEE S, KIM D. Deep learning based recommender system using cross convolutional filters[J]. Information sciences: an international journal, 2022, 592(C): 112–122. [13] 马甜甜, 杨长春, 严鑫杰, 等. 融合知识图谱和轻量级图卷积网络推荐系统的研究[J]. 智能系统学报, 2022, 17(4): 721–727. doi: 10.11992/tis.202107016 MA Tiantian, YANG Changchun, YAN Xinjie, et al. Research on the fusion of knowledge graph and lightweight graph convolutional network recommendation system[J]. CAAI transactions on intelligent systems, 2022, 17(4): 721–727. doi: 10.11992/tis.202107016 [14] CHEN Xuxin, WANG Ximin, ZHANG Ke, et al. Recent advances and clinical applications of deep learning in medical image analysis[J]. Medical image analysis, 2022, 79: 102444. doi: 10.1016/j.media.2022.102444 [15] 曾煜妮. 面向药物−靶标相互作用预测的多特征学习方法[D]. 成都: 四川大学, 2021. ZENG Yuni. Multi-feature learning approaches for drug-target interaction prediction[D]. Chengdu: Sichuan University, 2021. [16] JUMPER J, EVANS R, PRITZEL A, et al. Highly accurate protein structure prediction with AlphaFold[J]. Nature, 2021, 596(7873): 583–589. doi: 10.1038/s41586-021-03819-2 [17] RANA M M, NGUYEN D D. Geometric graph learning with extended atom-types features for protein-ligand binding affinity prediction[EB/OL]. (2023–01–15)[2023–08–09]. http://arxiv.org/abs/2301.06194. [18] MOESSER M A, KLEIN D K, BOYLES F, et al. Protein-ligand interaction graphs: learning from ligand-shaped 3D interaction graphs to improve binding affinity prediction[EB/OL]. (2022–03–07) [2023–08–09]. https://api.semanticscholar.org/CorpusID:247320144. [19] SURULIANDI A, IDHAYA T, RAJA S P. Drug target interaction prediction using machine learning techniques-A review[C]// 2021 Tenth International Conference on Intelligent Computing and Information Systems. Cairo: IEEE. [20] DHAKAL A, MCKAY C, TANNER J J, et al. Artificial intelligence in the prediction of protein-ligand interactions: recent advances and future directions[J]. Briefings in bioinformatics, 2022, 23(1): bbab476. doi: 10.1093/bib/bbab476 [21] ABBASI K, RAZZAGHI P, POSO A, et al. Deep learning in drug target interaction prediction: current and future perspectives[J]. Current medicinal chemistry, 2021, 28(11): 2100–2113. [22] SAMUEL C. Drug repositioning and indication discovery using description logics[D]. Cambridge: University of Cambridge, 2014. [23] ACEBRÓN-GARCÍA-DE-EULATE M, BLUNDELL T L, VEDITHI S C. Strategies for drug target identification in Mycobacterium leprae[J]. Drug discovery today, 2021, 26(7): 1569–1573. doi: 10.1016/j.drudis.2021.03.026 [24] TANOLI Z, SEEMAB U, SCHERER A, et al. Exploration of databases and methods supporting drug repurposing: a comprehensive survey[J]. Briefings in bioinformatics, 2021, 22(2): 1656–1678. doi: 10.1093/bib/bbaa003 [25] WANG Chen, KURGAN L. Survey of similarity-based prediction of drug-protein interactions[J]. Current medicinal chemistry, 2020, 27(35): 5856–5886. doi: 10.2174/0929867326666190808154841 [26] LIM S, LU Y, CHO C Y, et al. A review on compound-protein interaction prediction methods: data, format, representation and model[J]. Computational and structural biotechnology journal, 2021, 19: 1541–1556. doi: 10.1016/j.csbj.2021.03.004 [27] BAGHERIAN M, SABETI E, WANG Kai, et al. Machine learning approaches and databases for prediction of drug-target interaction: a survey paper[J]. Briefings in bioinformatics, 2021, 22(1): 247–269. doi: 10.1093/bib/bbz157 [28] RU Xiaoqing, YE Xiucai, SAKURAI T, et al. Current status and future prospects of drug-target interaction prediction[J]. Briefings in functional genomics, 2021, 20(5): 312–322. doi: 10.1093/bfgp/elab031 [29] NOSENGO N. New tricks for old drugs[J]. Nature, 2016, 534(7607): 314–316. doi: 10.1038/534314a [30] GAUDELET T, DAY B, JAMASB A R, et al. Utilizing graph machine learning within drug discovery and development[J]. Briefings in bioinformatics, 2021, 22(6): bbab159. doi: 10.1093/bib/bbab159 [31] JIN Guangxu, WONG S T C. Toward better drug repositioning: prioritizing and integrating existing methods into efficient pipelines[J]. Drug discovery today, 2014, 19(5): 637–644. doi: 10.1016/j.drudis.2013.11.005 [32] PUSHPAKOM S, IORIO F, EYERS P A, et al. Drug repurposing: progress, challenges and recommendations[J]. Nature reviews drug discovery, 2019, 18(1): 41–58. doi: 10.1038/nrd.2018.168 [33] SARDANA D, ZHU Cheng, ZHANG Minlu, et al. Drug repositioning for orphan diseases[J]. Briefings in bioinformatics, 2011, 12(4): 346–356. doi: 10.1093/bib/bbr021 [34] 李擎宇, 张孝昌, 王升启. 人工智能预测药物−靶标相互作用研究进展[J]. 中国药理学与毒理学杂志, 2022, 8(1): 1–10. LI Qingyu, ZHANG Xiaochang, WANG Shengqi. Research progress in artificial intelligence for predicting drug-target interactions[J]. Chinese journal of pharmacology and toxicology, 2022, 8(1): 1–10. [35] ZOU Yurong, WANG Ruihan, DU Meng, et al. Identifying protein-ligand interactions via a novel distance self-feedback biomolecular interaction network[J]. The journal of physical chemistry B, 2023, 127(4): 899–911. doi: 10.1021/acs.jpcb.2c07592 [36] SHEN Chao, HU Ye, WANG Zhe, et al. Can machine learning consistently improve the scoring power of classical scoring functions? Insights into the role of machine learning in scoring functions[J]. Briefings in bioinformatics, 2021, 22(1): 497–514. doi: 10.1093/bib/bbz173 [37] BALLESTER P J, MITCHELL J B O. A machine learning approach to predicting protein-ligand binding affinity with applications to molecular docking[J]. Bioinformatics, 2010, 26(9): 1169–1175. doi: 10.1093/bioinformatics/btq112 [38] HU Zhiqiang, LIU Wenfeng, ZHANG Chenbin, et al. SAM-DTA: a sequence-agnostic model for drug-target binding affinity prediction[J]. Briefings in bioinformatics, 2023, 24(1): bbac533. doi: 10.1093/bib/bbac533 [39] WEN Ming, ZHANG Zhimin, NIU Shaoyu, et al. Deep-learning-based drug-target interaction prediction[J]. Journal of proteome research, 2017, 16(4): 1401–1409. doi: 10.1021/acs.jproteome.6b00618 [40] PAHIKKALA T, AIROLA A, PIETILÄ S, et al. Toward more realistic drug-target interaction predictions[J]. Briefings in bioinformatics, 2015, 16(2): 325–337. doi: 10.1093/bib/bbu010 [41] RU Xiaoqing, YE Xiucai, SAKURAI T, et al. NerLTR-DTA: drug-target binding affinity prediction based on neighbor relationship and learning to rank[J]. Bioinformatics, 2022, 38(7): 1964–1971. doi: 10.1093/bioinformatics/btac048 [42] PLIAKOS K, VENS C, TSOUMAKAS G. Predicting drug-target interactions with multi-label classification and label partitioning[J]. IEEE/ACM transactions on computational biology and bioinformatics, 2021, 18(4): 1596–1607. doi: 10.1109/TCBB.2019.2951378 [43] CHU Yanyi, SHAN Xiaoqi, CHEN Tianhang, et al. DTI-MLCD: predicting drug-target interactions using multi-label learning with community detection method[J]. Briefings in bioinformatics, 2021, 22(3): bbaa205. doi: 10.1093/bib/bbaa205 [44] ZHENG Liangzhen, FAN Jingrong, MU Yuguang. OnionNet: a multiple-layer intermolecular-contact-based convolutional neural network for protein-ligand binding affinity prediction[J]. ACS omega, 2019, 4(14): 15956–15965. doi: 10.1021/acsomega.9b01997 [45] WANG D D, CHAN M T. Protein-ligand binding affinity prediction based on profiles of intermolecular contacts[J]. Computational and structural biotechnology journal, 2022, 20: 1088–1096. doi: 10.1016/j.csbj.2022.02.004 [46] SÁNCHEZ-CRUZ N, MEDINA-FRANCO J L, MESTRES J, et al. Extended connectivity interaction features: improving binding affinity prediction through chemical description[J]. Bioinformatics, 2021, 37(10): 1376–1382. doi: 10.1093/bioinformatics/btaa982 [47] KYRO G W, BRENT R I, BATISTA V S. HAC-net: a hybrid attention-based convolutional neural network for highly accurate protein-ligand binding affinity prediction[J]. Journal of chemical information and modeling, 2023, 63(7): 1947–1960. doi: 10.1021/acs.jcim.3c00251 [48] KWON Y, SHIN W H, KO J, et al. AK-score: accurate protein-ligand binding affinity prediction using an ensemble of 3D-convolutional neural networks[J]. International journal of molecular sciences, 2020, 21(22): 8424. doi: 10.3390/ijms21228424 [49] LI Yanjun, REZAEI M A, LI Chenglong, et al. DeepAtom: a framework for protein-ligand binding affinity prediction[C]//2019 IEEE International Conference on Bioinformatics and Biomedicine. San Diego: IEEE, 2019: 303–310. [50] WANG Yuxiao, QIU Zongzhao, JIAO Qihong, et al. Structure-based protein-drug affinity prediction with spatial attention mechanisms[C]//2021 IEEE International Conference on Bioinformatics and Biomedicine. Houston: IEEE, 2021: 92–97. [51] JIMÉNEZ J, ŠKALIČ M, MARTÍNEZ-ROSELL G, et al. Kdeep: protein-ligand absolute binding affinity prediction via 3D-convolutional neural networks[J]. Journal of chemical information and modeling, 2018, 58(2): 287–296. doi: 10.1021/acs.jcim.7b00650 [52] STEPNIEWSKA-DZIUBINSKA M M, ZIELENKIEWICZ P, SIEDLECKI P. Development and evaluation of a deep learning model for protein-ligand binding affinity prediction[J]. Bioinformatics, 2018, 34(21): 3666–3674. doi: 10.1093/bioinformatics/bty374 [53] WANG Yeji, WU Shuo, DUAN Yanwen, et al. A point cloud-based deep learning strategy for protein-ligand binding affinity prediction[J]. Briefings in bioinformatics, 2022, 23(1): bbab474. doi: 10.1093/bib/bbab474 [54] PANDEY M, RADAEVA M, MSLATI H, et al. Ligand binding prediction using protein structure graphs and residual graph attention networks[J]. Molecules, 2022, 27(16): 5114. doi: 10.3390/molecules27165114 [55] JIANG Dejun, HSIEH C Y, WU Zhenxing, et al. InteractionGraphNet: a novel and efficient deep graph representation learning framework for accurate protein-ligand interaction predictions[J]. Journal of medicinal chemistry, 2021, 64(24): 18209–18232. doi: 10.1021/acs.jmedchem.1c01830 [56] VOLKOV M, TURK J A, DRIZARD N, et al. On the frustration to predict binding affinities from protein-ligand structures with deep neural networks[J]. Journal of medicinal chemistry, 2022, 65(11): 7946–7958. doi: 10.1021/acs.jmedchem.2c00487 [57] ZHOU Jingbo, LI Shuangli, HUANG Liang, et al. Distance-aware molecule graph attention network for drug-target binding affinity prediction[EB/OL]. (2020–12–17)[2023–08–19]. http://arxiv.org/abs/2012.09624. [58] LI Shuangli, ZHOU Jingbo, XU Tong, et al. Structure-aware interactive graph neural networks for the prediction of protein-ligand binding affinity[C]//Proceedings of the 27th ACM SIGKDD Conference on Knowledge Discovery & Data Mining. Virtual Event, Singapore: ACM, 2021: 975–985. [59] LIM J, RYU S, PARK K, et al. Predicting drug-target interaction using a novel graph neural network with 3D structure-embedded graph representation[J]. Journal of chemical information and modeling, 2019, 59(9): 3981–3988. doi: 10.1021/acs.jcim.9b00387 [60] MOON S, ZHUNG W, YANG S, et al. PIGNet: a physics-informed deep learning model toward generalized drug-target interaction predictions[J]. Chemical science, 2022, 13(13): 3661–3673. doi: 10.1039/D1SC06946B [61] XIA Chunqiu, FENG Shihao, XIA Ying, et al. Leveraging scaffold information to predict protein-ligand binding affinity with an empirical graph neural network[J]. Briefings in bioinformatics, 2023, 24(1): bbac603. doi: 10.1093/bib/bbac603 [62] ZHANG Shuke, JIN Yanzhao, LIU Tianmeng, et al. SS-GNN: a simple-structured graph neural network for affinity prediction[J]. ACS omega, 2023, 8(25): 22496–22507. doi: 10.1021/acsomega.3c00085 [63] ZHANG Li, WANG Chunchun, CHEN Xing. Predicting drug-target binding affinity through molecule representation block based on multi-head attention and skip connection[J]. Briefings in bioinformatics, 2022, 23(6): bbac468. doi: 10.1093/bib/bbac468 [64] RIFAIOGLU A S, NALBAT E, ATALAY V, et al. DEEPScreen: high performance drug-target interaction prediction with convolutional neural networks using 2-D structural compound representations[J]. Chemical science, 2020, 11(9): 2531–2557. doi: 10.1039/C9SC03414E [65] MIKOLOV T, CHEN Kai, CORRADO G, et al. Efficient estimation of word representations in vector space[EB/OL]. (2013–01–16)[2023–08–19]. http://arxiv.org/abs/1301.3781. [66] HUANG Kexin, XIAO Cao, GLASS L M, et al. MolTrans: molecular interaction Transformer for drug-target interaction prediction[J]. Bioinformatics, 2021, 37(6): 830–836. doi: 10.1093/bioinformatics/btaa880 [67] KRENN M, HÄSE F, NIGAM A, et al. Self-referencing embedded strings (SELFIES): a 100% robust molecular string representation[J]. Machine learning: science and technology, 2020, 1(4): 045024. doi: 10.1088/2632-2153/aba947 [68] NGUYEN T, LE Hang, QUINN T P, et al. GraphDTA: predicting drug-target binding affinity with graph neural networks[J]. Bioinformatics, 2021, 37(8): 1140–1147. doi: 10.1093/bioinformatics/btaa921 [69] KARIMI M, WU Di, WANG Zhangyang, et al. DeepAffinity: interpretable deep learning of compound-protein affinity through unified recurrent and convolutional neural networks[J]. Bioinformatics, 2019, 35(18): 3329–3338. doi: 10.1093/bioinformatics/btz111 [70] SHARMA A, LYONS J, DEHZANGI A, et al. A feature extraction technique using bi-gram probabilities of position specific scoring matrix for protein fold recognition[J]. Journal of theoretical biology, 2013, 320: 41–46. doi: 10.1016/j.jtbi.2012.12.008 [71] SHEN Hongbin, CHOU Kuochen. Nuc-PLoc: a new web-server for predicting protein subnuclear localization by fusing PseAA composition and PsePSSM[J]. Protein engineering, design & selection: PEDS, 2007, 20(11): 561–567. [72] CHOU Kuochen. Using amphiphilic pseudo amino acid composition to predict enzyme subfamily classes[J]. Bioinformatics, 2005, 21(1): 10–19. doi: 10.1093/bioinformatics/bth466 [73] MAHMUD S M H, CHEN Wenyu, LIU Yongsheng, et al. PreDTIs: prediction of drug-target interactions based on multiple feature information using gradient boosting framework with data balancing and feature selection techniques[J]. Briefings in bioinformatics, 2021, 22(5): bbab046. doi: 10.1093/bib/bbab046 [74] WEE J, XIA Kelin. Forman persistent Ricci curvature (FPRC)-based machine learning models for protein-ligand binding affinity prediction[J]. Briefings in bioinformatics, 2021, 22(6): bbab136. doi: 10.1093/bib/bbab136 [75] MENG Zhenyu, XIA Kelin. Persistent spectral-based machine learning (PerSpect ML) for protein-ligand binding affinity prediction[J]. Science advances, 2021, 7(19): eabc5329. doi: 10.1126/sciadv.abc5329 [76] LIU Xiang, FENG Huitao, WU Jie, et al. Persistent spectral hypergraph based machine learning (PSH-ML) for protein-ligand binding affinity prediction[J]. Briefings in bioinformatics, 2021, 22(5): bbab127. doi: 10.1093/bib/bbab127 [77] REDKAR S, MONDAL S, JOSEPH A, et al. A machine learning approach for Drug-target interaction prediction using wrapper feature selection and class balancing[J]. Molecular informatics, 2020, 39(5): e1900062. doi: 10.1002/minf.201900062 [78] HE Tong, HEIDEMEYER M, BAN Fuqiang, et al. SimBoost: a read-across approach for predicting drug-target binding affinities using gradient boosting machines[J]. Journal of cheminformatics, 2017, 9(1): 24. doi: 10.1186/s13321-017-0209-z [79] ZENG Xiangxiang, ZHU Siyi, HOU Yuan, et al. Network-based prediction of drug-target interactions using an arbitrary-order proximity embedded deep forest[J]. Bioinformatics, 2020, 36(9): 2805–2812. doi: 10.1093/bioinformatics/btaa010 [80] CHEN Cheng, SHI Han, JIANG Zhiwen, et al. DNN-DTIs: improved drug-target interactions prediction using XGBoost feature selection and deep neural network[J]. Computers in biology and medicine, 2021, 136: 104676. doi: 10.1016/j.compbiomed.2021.104676 [81] LI Zhanchao, HUANG Menghua, ZHONG Wenqian, et al. Identification of drug-target interaction from interactome network with ‘guilt-by-association’ principle and topology features[J]. Bioinformatics, 2016, 32(7): 1057–1064. doi: 10.1093/bioinformatics/btv695 [82] TANOORI B, JAHROMI M Z. Using drug-drug and protein-protein similarities as feature vector for drug-target binding prediction[J]. Chemometrics and intelligent laboratory systems, 2021, 217: 104405. doi: 10.1016/j.chemolab.2021.104405 [83] TANOORI B, JAHROMI M Z, MANSOORI E G. Drug-target continuous binding affinity prediction using multiple sources of information[J]. Expert systems with applications, 2021, 186: 115810. doi: 10.1016/j.eswa.2021.115810 [84] CHEN Zhanheng, YOU Zhuhong, GUO Zhenhao, et al. Prediction of drug-target interactions from multi-molecular network based on deep walk embedding model[J]. Frontiers in bioengineering and biotechnology, 2020, 8: 338. doi: 10.3389/fbioe.2020.00338 [85] XUAN Ping, CHEN Bingxu, ZHANG Tiangang, et al. Prediction of drug–target interactions based on network representation learning and ensemble learning[J]. IEEE/ACM transactions on computational biology and bioinformatics, 2021, 18(6): 2671–2681. doi: 10.1109/TCBB.2020.2989765 [86] CHU Yanyi, KAUSHIK A C, WANG Xiangeng, et al. DTI-CDF: a cascade deep forest model towards the prediction of drug-target interactions based on hybrid features[J]. Briefings in bioinformatics, 2021, 22(1): 451–462. doi: 10.1093/bib/bbz152 [87] LIU Ran, LIU Xiang, WU Jie. Persistent path-spectral (PPS) based machine learning for protein-ligand binding affinity prediction[J]. Journal of chemical information and modeling, 2023, 63(3): 1066–1075. doi: 10.1021/acs.jcim.2c01251 [88] VASWANI A, SHAZEER N, PARMAR N, et al. Attention is all you need[C]//Proceedings of the 31st International Conference on Neural Information Processing Systems. California: ACM, 2017: 6000–6010. [89] WANG Xianfang, LIU Yifeng, LU Fan, et al. Dipeptide frequency of word frequency and graph convolutional networks for DTA prediction[J]. Frontiers in bioengineering and biotechnology, 2020, 8: 267. doi: 10.3389/fbioe.2020.00267 [90] LIN Xuan. DeepGS: deep representation learning of graphs and sequences for drug-target binding affinity prediction[EB/OL]. (2020–03–31)[2023–08–19]. http://arxiv.org/abs/2003.13902. [91] WANG Shudong, DU Zhenzhen, DING Mao, et al. LDCNN-DTI: a novel light deep convolutional neural network for drug-target interaction predictions[C]//2020 IEEE International Conference on Bioinformatics and Biomedicine. Seoul: IEEE, 2020: 1132–1136. [92] AGYEMANG B, WU Weiping, KPIEBAAREH M Y, et al. Multi-view self-attention for interpretable drug-target interaction prediction[J]. Journal of biomedical informatics, 2020, 110: 103547. doi: 10.1016/j.jbi.2020.103547 [93] JIANG Mingjian, LI Zhen, ZHANG Shugang, et al. Drug-target affinity prediction using graph neural network and contact maps[J]. RSC advances, 2020, 10(35): 20701–20712. doi: 10.1039/D0RA02297G [94] KAO Poyu, KAO Shumin, HUANG Nanlan, et al. Toward drug-target interaction prediction via ensemble modeling and transfer learning[C]//2021 IEEE International Conference on Bioinformatics and Biomedicine. Houston: IEEE, 2021: 2384–2391. [95] WU Yifan, GAO Min, ZENG Min, et al. BridgeDPI: a novel graph neural network for predicting drug-protein interactions[J]. Bioinformatics, 2022, 38(9): 2571–2578. doi: 10.1093/bioinformatics/btac155 [96] 王红梅, 郭真俊, 张丽杰. 基于图神经网络的药物−靶标相互作用预测研究[J]. 长春工业大学学报, 2021, 42(4): 318–325. WANG Hongmei, GUO Zhenjun, ZHANG Lijie. Drug-target interaction prediction based on graph neural network[J]. Journal of Changchun university of technology, 2021, 42(4): 318–325. [97] CHEN Ruolan, XIA Feng, HU Bing, et al. Drug-target interactions prediction via deep collaborative filtering with multiembeddings[J]. Briefings in bioinformatics, 2022, 23(2): bbab520. doi: 10.1093/bib/bbab520 [98] CHEN Jiatao, ZHANG Liang, CHENG Ke, et al. Exploring multi-level mutual information for drug-target interaction prediction[C]//2020 IEEE International Conference on Bioinformatics and Biomedicine. Seoul: IEEE, 2020: 251–256. [99] PENG Jiajie, WANG Yuxian, GUAN Jiaojiao, et al. An end-to-end heterogeneous graph representation learning-based framework for drug-target interaction prediction[J]. Briefings in bioinformatics, 2021, 22(5): bbaa430. doi: 10.1093/bib/bbaa430 [100] LI Yuhui, LIANG Wei, PENG Li, et al. Predicting drug-target interactions via dual-stream graph neural network[J]. IEEE/ACM transactions on computational biology and bioinformatics, 2022: 3204188. . [101] ZHAO Chengshuai, LIU Shuai, HUANG Feng, et al. CSGNN: contrastive self-supervised graph neural network for molecular interaction prediction[C]//Proceeding of the 30th International Joint Conference on Artificial Intelligence. 2021: 3756–3763. [102] ZENG Yuni, CHEN Xiangru, LUO Yujie, et al. Deep drug-target binding affinity prediction with multiple attention blocks[J]. Briefings in bioinformatics, 2021, 22(5): bbab117. doi: 10.1093/bib/bbab117 [103] ALEB N. A mutual attention model for drug target binding affinity prediction[J]. IEEE/ACM transactions on computational biology and bioinformatics, 2022, 19(6): 3224–3232. [104] GAO K Y, FOKOUE A, LUO Heng, et al. Interpretable drug target prediction using deep neural representation[C]//Proceeding of the 27th International Joint Conference on Artificial Intelligence. Stockholm: AAAI Press, 2018: 3371–3377. [105] ABBASI K, RAZZAGHI P, POSO A, et al. DeepCDA: deep cross-domain compound-protein affinity prediction through LSTM and convolutional neural networks[J]. Bioinformatics, 2020, 36(17): 4633–4642. doi: 10.1093/bioinformatics/btaa544 [106] KARIMI M, WU D, WANG Z, et al. Explainable deep relational networks for predicting compound-protein affinities and contacts[J]. Journal of chemical information and modeling, 2021, 61(1): 46–66. doi: 10.1021/acs.jcim.0c00866 [107] ZHAO Qichang, ZHAO Haochen, ZHENG Kai, et al. HyperAttentionDTI: improving drug-protein interaction prediction by sequence-based deep learning with attention mechanism[J]. Bioinformatics, 2022, 38(3): 655–662. doi: 10.1093/bioinformatics/btab715 [108] CHEN Lifan, TAN Xiaoqin, WANG Dingyan, et al. TransformerCPI: improving compound-protein interaction prediction by sequence-based deep learning with self-attention mechanism and label reversal experiments[J]. Bioinformatics, 2020, 36(16): 4406–4414. doi: 10.1093/bioinformatics/btaa524 [109] BAI Peizhen, MILJKOVIĆ F, JOHN B, et al. Interpretable bilinear attention network with domain adaptation improves drug-target prediction[J]. Nature machine intelligence, 2023, 5: 126–136. doi: 10.1038/s42256-022-00605-1 [110] CHENG Zhongjian, ZHAO Qichang, LI Yaohang, et al. IIFDTI: predicting drug-target interactions through interactive and independent features based on attention mechanism[J]. Bioinformatics, 2022, 38(17): 4153–4161. doi: 10.1093/bioinformatics/btac485 [111] ZHAO Qichang, DUAN Guihua, YANG Mengyun, et al. AttentionDTA: drug-target binding affinity prediction by sequence-based deep learning with attention mechanism[J]. IEEE/ACM transactions on computational biology and bioinformatics, 2023, 20(2): 852–863. doi: 10.1109/TCBB.2022.3170365 [112] HUANG Lei, LIN Jiecong, LIU Rui, et al. CoaDTI: multi-modal co-attention based framework for drug-target interaction annotation[J]. Briefings in bioinformatics, 2022, 23(6): bbac446. doi: 10.1093/bib/bbac446 [113] WANG Tianyu, YANG Wenming, CHEN Jie, et al. ConformerDTI: local features coupling global representations for drug-target interaction prediction[C]//2022 IEEE International Conference on Bioinformatics and Biomedicine. Las Vegas: IEEE, 2022: 1227–1234. [114] LI Fei, ZHANG Ziqiao, GUAN Jihong, et al. Effective drug-target interaction prediction with mutual interaction neural network[J]. Bioinformatics, 2022, 38(14): 3582–3589. doi: 10.1093/bioinformatics/btac377 [115] MONTEIRO N R C, OLIVEIRA J L, ARRAIS J P. DTITR: end-to-end drug-target binding affinity prediction with transformers[J]. Computers in biology and medicine, 2022, 147: 105772. doi: 10.1016/j.compbiomed.2022.105772 [116] TSUBAKI M, TOMII K, JUN Sese. Compound-protein interaction prediction with end-to-end learning of neural networks for graphs and sequences[J]. Bioinformatics, 2019, 35(2): 309–318. doi: 10.1093/bioinformatics/bty535 [117] YANG Ziduo, ZHONG Weihe, ZHAO Lu, et al. ML-DTI: mutual learning mechanism for interpretable drug-target interaction prediction[J]. The journal of physical chemistry letters, 2021, 12(17): 4247–4261. doi: 10.1021/acs.jpclett.1c00867 [118] CHENG Zhongjian, YAN Cheng, WU Fangxiang, et al. Drug-target interaction prediction using multi-head self-attention and graph attention network[J]. IEEE/ACM transactions on computational biology and bioinformatics, 2022, 19(4): 2208–2218. doi: 10.1109/TCBB.2021.3077905 [119] YUAN Weining, CHEN Guanxing, CHEN C Y C. FusionDTA: attention-based feature polymerizer and knowledge distillation for drug-target binding affinity prediction[J]. Briefings in bioinformatics, 2022, 23(1): bbab506. doi: 10.1093/bib/bbab506 [120] NGUYEN T M, NGUYEN T, LE T M, et al. GEFA: early fusion approach in drug-target affinity prediction[J]. IEEE/ACM trans comput biol bioinform, 2022, 19(2): 718–728. doi: 10.1109/TCBB.2021.3094217 [121] CHU Zhaoyang, HUANG Feng, FU Haitao, et al. Hierarchical graph representation learning for the prediction of drug-target binding affinity[J]. Information sciences: an international journal, 2022, 613(C): 507–523. [122] ZHAO Tianyi, HU Yang, VALSDOTTIR L R, et al. Identifying drug-target interactions based on graph convolutional network and deep neural network[J]. Briefings in bioinformatics, 2021, 22(2): 2141–2150. doi: 10.1093/bib/bbaa044 [123] FU Haitao, HUANG Feng, LIU Xuan, et al. MVGCN: data integration through multi-view graph convolutional network for predicting links in biomedical bipartite networks[J]. Bioinformatics, 2022, 38(2): 426–434. doi: 10.1093/bioinformatics/btab651 [124] LI Chunyan, YAO Junfeng, WEI Wei, et al. Geometry-based molecular generation with deep constrained variational autoencoder[J]. IEEE transactions on neural networks and learning systems, 2024, 35(4): 4852–4861. doi: 10.1109/TNNLS.2022.3147790 [125] LI Jiahan. Directed weight neural networks for protein structure representation learning[EB/OL]. (2022–01–28)[2023–08–19]. http://arxiv.org/abs/2201.13299. [126] SON J, KIM D. Development of a graph convolutional neural network model for efficient prediction of protein-ligand binding affinities[J]. PLoS One, 2021, 16(4): e0249404. doi: 10.1371/journal.pone.0249404 [127] SEO S, CHOI J, PARK S, et al. Binding affinity prediction for protein-ligand complex using deep attention mechanism based on intermolecular interactions[J]. BMC bioinformatics, 2021, 22(1): 542. doi: 10.1186/s12859-021-04466-0 [128] CANG Zixuan, MU Lin, WEI Guowei. Representability of algebraic topology for biomolecules in machine learning based scoring and virtual screening[J]. PLoS computational biology, 2018, 14(1): e1005929. doi: 10.1371/journal.pcbi.1005929 [129] NGUYEN D D, GAO Kaifu, WANG Menglun, et al. MathDL: mathematical deep learning for D3R grand challenge 4[J]. Journal of computer-aided molecular design, 2020, 34(2): 131–147. doi: 10.1007/s10822-019-00237-5 [130] JIANG Peiran, CHI Ying, LI Xiaoshuang, et al. Molecular persistent spectral image (Mol-PSI) representation for machine learning models in drug design[J]. Briefings in bioinformatics, 2022, 23(1): bbab527. doi: 10.1093/bib/bbab527 [131] BREIMAN L. Random forests[J]. Machine learning, 2001, 45: 5–32. doi: 10.1023/A:1010933404324 [132] FRIEDMAN J H. Greedy function approximation: a gradient boosting machine[J]. The annals of statistics, 2001, 29(5): 1189–1232. doi: 10.1214/aos/1013203450 [133] 叶贵鑫, 张宇翔, 张成, 等. 基于图神经网络的OpenCL程序自动优化启发式方法[J]. 计算机研究与发展, 2023, 60(5): 1121–1135. doi: 10.7544/issn1000-1239.202110943 YE Guixin, ZHANG Yuxiang, ZHANG Cheng, et al. Automatic optimization heuristics method for OpenCL program based on graph neural network[J]. Journal of computer research and development, 2023, 60(5): 1121–1135. doi: 10.7544/issn1000-1239.202110943 [134] YI Yiqiang, WAN Xu, ZHAO Kangfei, et al. Predicting protein-ligand binding affinity with equivariant line graph network[EB/OL]. (2022–10–27)[2023–08–19]. http://arxiv.org/abs/2210.16098. [135] CHEN Peng, SHEN Huimin, ZHANG Youzhi, et al. SGNet: sequence-based convolution and ligand graph network for protein binding affinity prediction[J]. IEEE/ACM transactions on computational biology and bioinformatics, 2023, 20(5): 3257–3266. doi: 10.1109/TCBB.2023.3262821 [136] LI Xiaoshuang, LIU Xiang, LU Le, et al. Multiphysical graph neural network (MP-GNN) for COVID-19 drug design[J]. Briefings in bioinformatics, 2022, 23(4): bbac231. doi: 10.1093/bib/bbac231 [137] ZHAO Bowei, YOU Zhuhong, HU Lun, et al. A novel method to predict drug-target interactions based on large-scale graph representation learning[J]. Cancers, 2021, 13(9): 2111. doi: 10.3390/cancers13092111 [138] SHEN Ying, ZHANG Yilin, YUAN Kaiqi, et al. A knowledge-enhanced multi-view framework for drug-target interaction prediction[J]. IEEE transactions on big data, 2022, 8(5): 1387–1398. doi: 10.1109/TBDATA.2021.3051673 [139] YU Zhimiao, LU Jiarui, JIN Yuan, et al. KenDTI: an ensemble model for predicting drug-target interaction by integrating multi-source information[J]. IEEE/ACM transactions on computational biology and bioinformatics, 2021, 18(4): 1305–1314. doi: 10.1109/TCBB.2021.3074401 [140] HINNERICHS T, HOEHNDORF R. DTI-Voodoo: machine learning over interaction networks and ontology-based background knowledge predicts drug-target interactions[J]. Bioinformatics, 2021, 37(24): 4835–4843. doi: 10.1093/bioinformatics/btab548 [141] PU Yuqian, LI Jiawei, TANG Jijun, et al. DeepFusionDTA: drug-target binding affinity prediction with information fusion and hybrid deep-learning ensemble model[J]. IEEE/ACM transactions on computational biology and bioinformatics, 2022, 19(5): 2760–2769. doi: 10.1109/TCBB.2021.3103966 [142] CHEN Jiatao, ZHANG Liang, CHENG Ke, et al. Predicting drug-target interaction via self-supervised learning[J]. IEEE/ACM transactions on computational biology and bioinformatics, 2023, 20(5): 2781–2789. [143] JONES D, KIM H, ZHANG Xiaohua, et al. Improved protein-ligand binding affinity prediction with structure-based deep fusion inference[J]. Journal of chemical information and modeling, 2021, 61(4): 1583–1592. doi: 10.1021/acs.jcim.0c01306 [144] YELLA J K, GHANDIKOTA S K, JEGGA A G. GraMDTA: multimodal graph neural networks for predicting drug-target associations[C]//2022 IEEE International Conference on Bioinformatics and Biomedicine. Las Vegas: IEEE, 2022: 1957–1965. [145] LI Qingyu, ZHANG Xiaochang, WU Lianlian, et al. PLA-MoRe: a protein-ligand binding affinity prediction model via comprehensive molecular representations[J]. Journal of chemical information and modeling, 2022, 62(18): 4380–4390. doi: 10.1021/acs.jcim.2c00960 [146] GEURTS P, ERNST D, WEHENKEL L. Extremely randomized trees[J]. Machine learning, 2006, 63(1): 3–42. doi: 10.1007/s10994-006-6226-1 [147] KE Guolin, MENG Qi, FINLEY T, et al. LightGBM: a highly efficient gradient boosting decision tree[C]//Proceedings of the 31st International Conference on Neural Information Processing Systems. Long Beach: ACM, 2017: 3149–3157. [148] CHAN T F, GOLUB G H, LEVEQUE R J. Updating formulae and a pairwise algorithm for computing sample variances[C]//COMPSTAT 1982 5th Symposium held at Toulouse 1982. Heidelberg: Physica, 1982: 30–41. [149] PISNER D A, SCHNYER D M. Support vector machine[M]//Machine Learning. Amsterdam: Elsevier, 2020: 101–121. [150] SUN Chang, XUAN Ping, ZHANG Tiangang, et al. Graph convolutional autoencoder and generative adversarial network-based method for predicting drug-target interactions[J]. IEEE/ACM transactions on computational biology and bioinformatics, 2022, 19(1): 455–464. doi: 10.1109/TCBB.2020.2999084 [151] BAGHERIAN M, KIM R B, JIANG Cheng, et al. Coupled matrix-matrix and coupled tensor-matrix completion methods for predicting drug-target interactions[J]. Briefings in bioinformatics, 2021, 22(2): 2161–2171. doi: 10.1093/bib/bbaa025 [152] LI Jin, WANG Jingru, LYU Hao, et al. IMCHGAN: inductive matrix completion with heterogeneous graph attention networks for drug-target interactions prediction[J]. IEEE/ACM transactions on computational biology and bioinformatics, 2022, 19(2): 655–665. doi: 10.1109/TCBB.2021.3088614 [153] WANG Shiming, LI Jie, WANG Yadong, et al. A neighborhood-based global network model to predict drug-target interactions[J]. IEEE/ACM transactions on computational biology and bioinformatics, 2022, 19(4): 2017–2025. doi: 10.1109/TCBB.2021.3064614 [154] WANG Xiaoqi, YANG Yaning, LI Kenli, et al. BioERP: biomedical heterogeneous network-based self-supervised representation learning approach for entity relationship predictions[J]. Bioinformatics, 2021, 37(24): 4793–4800. doi: 10.1093/bioinformatics/btab565 [155] ZENG Xiangxiang, ZHU Siyi, LU Weiqiang, et al. Target identification among known drugs by deep learning from heterogeneous networks[J]. Chemical science, 2020, 11(7): 1775–1797. doi: 10.1039/C9SC04336E [156] SHANG Yifan, YE Xiucai, FUTAMURA Y, et al. Multiview network embedding for drug-target interactions prediction by consistent and complementary information preserving[J]. Briefings in bioinformatics, 2022, 23(3): bbac059. doi: 10.1093/bib/bbac059 [157] WAN Fangping, HONG Lixiang, XIAO An, et al. NeoDTI: neural integration of neighbor information from a heterogeneous network for discovering new drug-target interactions[J]. Bioinformatics, 2019, 35(1): 104–111. doi: 10.1093/bioinformatics/bty543 [158] LUO Yunan, ZHAO Xinbin, ZHOU Jingtian, et al. A network integration approach for drug-target interaction prediction and computational drug repositioning from heterogeneous information[J]. Nature communications, 2017, 8(1): 573. doi: 10.1038/s41467-017-00680-8 [159] ZHAO Lingling, WANG Junjie, PANG Long, et al. GANsDTA: predicting drug-target binding affinity using GANs[J]. Frontiers in genetics, 2020, 10: 1243. doi: 10.3389/fgene.2019.01243 [160] JIANG Lu, SUN Jiahao, WANG Yue, et al. Identifying drug-target interactions via heterogeneous graph attention networks combined with cross-modal similarities[J]. Briefings in bioinformatics, 2022, 23(2): bbac016. doi: 10.1093/bib/bbac016 [161] LI Mei, CAI Xiangrui, XU Sihan, et al. Metapath-aggregated heterogeneous graph neural network for drug-target interaction prediction[J]. Briefings in bioinformatics, 2023, 24(1): bbac578. doi: 10.1093/bib/bbac578 [162] WENG Yuyou, LIN Chen, ZENG Xiangxiang, et al. Drug target interaction prediction using Multi-task learning and Co-attention[C]//2019 IEEE International Conference on Bioinformatics and Biomedicine. San Diego: IEEE, 2019: 528–533. [163] WANG Hongzhun, HUANG Feng, XIONG Zhankun, et al. A heterogeneous network-based method with attentive meta-path extraction for predicting drug-target interactions[J]. Briefings in bioinformatics, 2022, 23(4): bbac184. doi: 10.1093/bib/bbac184 [164] SHANG Yifan, GAO Lin, ZOU Quan, et al. Prediction of drug-target interactions based on multi-layer network representation learning[J]. Neurocomputing, 2021, 434: 80–89. doi: 10.1016/j.neucom.2020.12.068 [165] XUAN Ping, ZHANG Yu, CUI Hui, et al. Integrating multi-scale neighbouring topologies and cross-modal similarities for drug-protein interaction prediction[J]. Briefings in bioinformatics, 2021, 22(5): bbab119. doi: 10.1093/bib/bbab119 [166] TORNG W, ALTMAN R B. Graph convolutional neural networks for predicting drug-target interactions[J]. Journal of chemical information and modeling, 2019, 59(10): 4131–4149. doi: 10.1021/acs.jcim.9b00628 [167] LI Tianjiao, ZHAO Xingming, LI Limin. Co-VAE: drug-target binding affinity prediction by co-regularized variational autoencoders[J]. IEEE transactions on pattern analysis and machine intelligence, 2022, 44(12): 8861–8873. doi: 10.1109/TPAMI.2021.3120428 [168] BROWN T B, MANN B, RYDER N, et al. Language models are few-shot learners[C]//Proceedings of the 34th International Conference on Neural Information Processing Systems. Vancouver: ACM, 2020: 1877–1901. [169] WANG Chunyu, ZHU Yan, WEN Naifeng, et al. SeqGO-CPA: improving compound-protein binding affinity prediction with sequence information and gene ontology knowledge[C]//2021 IEEE International Conference on Bioinformatics and Biomedicine. Houston: IEEE, 2021: 354–359. [170] NGUYEN T M, NGUYEN T, TRAN T. Mitigating cold-start problems in drug-target affinity prediction with interaction knowledge transferring[J]. Briefings in bioinformatics, 2022, 23(4): bbac269. doi: 10.1093/bib/bbac269 [171] LIU Shengchao, WANG Hanchen, LIU Weiyang, et al. Pre-training molecular graph representation with 3D geometry[EB/OL]. (2021–10–07)[2023–08–19]. http://arxiv.org/abs/2110.07728. [172] XIA Jun, ZHU Yanqiao, DU Yuanqi, et al. A systematic survey of chemical pre-trained models[C]//Proceedings of the Thirty-Second International Joint Conference on Artificial Intelligence. California: International Joint Conferences on Artificial Intelligence Organization, 2023: 6787–6795. [173] RIVES A, MEIER J, SERCU T, et al. Biological structure and function emerge from scaling unsupervised learning to 250 million protein sequences[J]. Proceedings of the national academy of sciences of the United States of America, 2021, 118(15): e2016239118. [174] LI Yang, QIAO Guanyu, GAO Xin, et al. Supervised graph co-contrastive learning for drug-target interaction prediction[J]. Bioinformatics, 2022, 38(10): 2847–2854. doi: 10.1093/bioinformatics/btac164 [175] LI Mei, XU Sihan, CAI Xiangrui, et al. Contrastive meta-learning for drug-target binding affinity prediction[C]//2022 IEEE International Conference on Bioinformatics and Biomedicine. Las Vegas: IEEE, 2022: 464–470. [176] ZHU Hui, YANG Jincai, HUANG Niu. Assessment of the generalization abilities of machine-learning scoring functions for structure-based virtual screening[J]. Journal of chemical information and modeling, 2022, 62(22): 5485–5502. doi: 10.1021/acs.jcim.2c01149 [177] LAMB J, CRAWFORD E D, PECK D, et al. The Connectivity Map: using gene-expression signatures to connect small molecules, genes, and disease[J]. Science, 2006, 313(5795): 1929–1935. doi: 10.1126/science.1132939 [178] ZITNIK M, NGUYEN F, WANG Bo, et al. Machine learning for integrating data in biology and medicine: principles, practice, and opportunities[J]. An international journal on information fusion, 2019, 50: 71–91. doi: 10.1016/j.inffus.2018.09.012 [179] DING Yijie, TANG Jijun, GUO Fei. Identification of drug–target interactions via fuzzy bipartite local model[J]. Neural computing and applications, 2020, 32(14): 10303–10319. doi: 10.1007/s00521-019-04569-z [180] DING Yijie, TANG Jijun, GUO Fei. Identification of drug-target interactions via dual Laplacian regularized least squares with multiple kernel fusion[J]. Knowledge based system, 2020, 204: 106254. doi: 10.1016/j.knosys.2020.106254 [181] CHEN Huiyuan, CHENG Feixiong, LI Jing. IDrug: integration of drug repositioning and drug-target prediction via cross-network embedding[J]. PLoS computational biology, 2020, 16(7): e1008040. doi: 10.1371/journal.pcbi.1008040 [182] DING Yijie, TANG Jijun, GUO Fei. Identification of drug-target interactions via multi-view graph regularized link propagation model[J]. Neurocomputing, 2021, 461: 618–631. doi: 10.1016/j.neucom.2021.05.100 [183] SUN Chang, CAO Yangkun, WEI Jinmao, et al. Autoencoder-based drug-target interaction prediction by preserving the consistency of chemical properties and functions of drugs[J]. Bioinformatics, 2021, 37(20): 3618–3625. doi: 10.1093/bioinformatics/btab384 [184] OLAYAN R S, ASHOOR H, BAJIC V B. DDR: efficient computational method to predict drug-target interactions using graph mining and machine learning approaches[J]. Bioinformatics, 2018, 34(7): 1164–1173. doi: 10.1093/bioinformatics/btx731 [185] XU Xiaoqiang, XUAN Ping, ZHANG Tiangang, et al. Inferring drug-target interactions based on random walk and convolutional neural network[J]. IEEE/ACM transactions on computational biology and bioinformatics, 2022, 19(4): 2294–2304. doi: 10.1109/TCBB.2021.3066813 [186] DING Yijie, TANG Jijun, GUO Fei, et al. Identification of drug-target interactions via multiple kernel-based triple collaborative matrix factorization[J]. Briefings in bioinformatics, 2022, 23(2): bbab582. doi: 10.1093/bib/bbab582 [187] TIAN Zhen, PENG Xiangyu, FANG Haichuan, et al. MHADTI: predicting drug-target interactions via multiview heterogeneous information network embedding with hierarchical attention mechanisms[J]. Briefings in bioinformatics, 2022, 23(6): bbac434. doi: 10.1093/bib/bbac434 [188] LIU Bin, PAPADOPOULOS D, MALLIAROS F D, et al. Multiple similarity drug-target interaction prediction with random walks and matrix factorization[J]. Briefings in bioinformatics, 2022, 23(5): bbac353. doi: 10.1093/bib/bbac353 [189] AN Qi, YU Liang. A heterogeneous network embedding framework for predicting similarity-based drug-target interactions[J]. Briefings in bioinformatics, 2021, 22(6): bbab275. doi: 10.1093/bib/bbab275 [190] HUANG Lan, LUO Huimin, LI Suning, et al. Drug-drug similarity measure and its applications[J]. Briefings in bioinformatics, 2021, 22(4): bbaa265. [191] WANG Bo, MEZLINI A M, DEMIR F, et al. Similarity network fusion for aggregating data types on a genomic scale[J]. Nature methods, 2014, 11(3): 333–337. doi: 10.1038/nmeth.2810 [192] PEROZZI B, AL-RFOU R, SKIENA S. DeepWalk: online learning of social representations[C]//Proceedings of the 20th ACM SIGKDD international conference on Knowledge discovery and data mining. New York: ACM, 2014: 701–710. [193] GROVER A, LESKOVEC J. node2vec: scalable feature learning for networks[J]. KDD: proceedings international conference on knowledge discovery & data mining, 2016, 2016: 855–864. [194] DONG Yuxiao, CHAWLA N V, SWAMI A. metapath2vec: scalable representation learning for heterogeneous networks[C]//Proceedings of the 23rd ACM SIGKDD International Conference on Knowledge Discovery and Data Mining. Halifax: ACM, 2017: 135–144. [195] LI Guodong, ZHANG Ping, SUN Weicheng, et al. Bridging-BPs: a novel approach to predict potential drug-target interactions based on a bridging heterogeneous graph and BPs2vec[J]. Briefings in bioinformatics, 2022, 23(2): bbab557. doi: 10.1093/bib/bbab557 [196] BORDES A, USUNIER N, GARCIA-DURÁN A, et al. Translating embeddings for modeling multi-relational data[C]//Proceedings of the 26th International Conference on Neural Information Processing Systems - Volume 2. Lake Tahoe: ACM, 2013: 2787–2795. [197] YANG Bishan, YIH W T, HE Xiaodong, et al. Embedding entities and relations for learning and inference in knowledge bases[EB/OL]. (2014–12–20)[2023–08–19]. http://arxiv.org/abs/1412.6575. [198] TROUILLON T, WELBL J, RIEDEL S, et al. Complex embeddings for simple link prediction[C]//Proceedings of the 33rd International Conference on International Conference on Machine Learning-Volume 48. New York: ACM, 2016: 2071–2080. [199] YE Qing, HSIEH C Y, YANG Ziyi, et al. A unified drug-target interaction prediction framework based on knowledge graph and recommendation system[J]. Nature communications, 2021, 12(1): 6775. doi: 10.1038/s41467-021-27137-3 [200] MOHAMED S K, NOUNU A, NOVÁČEK V. Drug target discovery using knowledge graph embeddings[C]//Proceedings of the 34th ACM/SIGAPP Symposium on Applied Computing. Limassol: ACM, 2019: 11–18. [201] MOHAMED S K, NOVÁČEK V, NOUNU A. Discovering protein drug targets using knowledge graph embeddings[J]. Bioinformatics, 2020, 36(2): 603–610. doi: 10.1093/bioinformatics/btz600 [202] SHAO Kanghao, ZHANG Yunhao, WEN Yuqi, et al. DTI-HETA: prediction of drug-target interactions based on GCN and GAT on heterogeneous graph[J]. Briefings in bioinformatics, 2022, 23(3): bbac109. doi: 10.1093/bib/bbac109 [203] LI Mei, CAI Xiangrui, LI Linyu, et al. Heterogeneous graph attention network for drug-target interaction prediction[C]//Proceedings of the 31st ACM International Conference on Information & Knowledge Management. Atlanta: ACM, 2022: 1166–1176. [204] LI Yang, QIAO Guanyu, WANG Keqi, et al. Drug-target interaction predication via multi-channel graph neural networks[J]. Briefings in bioinformatics, 2022, 23(1): bbab346. doi: 10.1093/bib/bbab346 [205] MENDEZ D, GAULTON A, BENTO A P, et al. ChEMBL: towards direct deposition of bioassay data[J]. Nucleic acids research, 2019, 47(D1): D930–D940. doi: 10.1093/nar/gky1075 [206] WISHART D S, FEUNANG Y D, GUO A C, et al. DrugBank 5.0: a major update to the DrugBank database for 2018[J]. Nucleic acids research, 2018, 46(D1): D1074–D1082. doi: 10.1093/nar/gkx1037 [207] SZKLARCZYK D, SANTOS A, VON MERING C, et al. STITCH 5: augmenting protein-chemical interaction networks with tissue and affinity data[J]. Nucleic acids research, 2016, 44(D1): D380–D384. doi: 10.1093/nar/gkv1277 [208] GILSON M K, LIU Tiqing, BAITALUK M, et al. BindingDB in 2015: a public database for medicinal chemistry, computational chemistry and systems pharmacology[J]. Nucleic acids research, 2016, 44(D1): D1045–D1053. doi: 10.1093/nar/gkv1072 [209] LIU Zhihai, SU Minyi, HAN Li, et al. Forging the basis for developing protein-ligand interaction scoring functions[J]. Accounts of chemical research, 2017, 50(2): 302–309. doi: 10.1021/acs.accounts.6b00491 [210] STATHIAS V, TURNER J, KOLETI A, et al. LINCS Data Portal 2.0: next generation access point for perturbation-response signatures[J]. Nucleic acids research, 2020, 48(D1): D431–D439. doi: 10.1093/nar/gkz1023 [211] HARDING S D, ARMSTRONG J F, FACCENDA E, et al. The IUPHAR/BPS guide to PHARMACOLOGY in 2022: curating pharmacology for COVID-19, malaria and antibacterials[J]. Nucleic acids research, 2022, 50(D1): D1282–D1294. doi: 10.1093/nar/gkab1010 [212] DEL TORO N, SHRIVASTAVA A, RAGUENEAU E, et al. The IntAct database: efficient access to fine-grained molecular interaction data[J]. Nucleic acids research, 2022, 50(D1): D648–D653. doi: 10.1093/nar/gkab1006 [213] FRESHOUR S L, KIWALA S, COTTO K C, et al. Integration of the Drug-Gene interaction database (DGIdb 4.0) with open crowdsource efforts[J]. Nucleic acids research, 2021, 49(D1): D1144–D1151. doi: 10.1093/nar/gkaa1084 [214] GALLO K, GOEDE A, ECKERT A, et al. PROMISCUOUS 2.0: a resource for drug-repositioning[J]. Nucleic acids research, 2021, 49(D1): D1373–D1380. doi: 10.1093/nar/gkaa1061 [215] TANOLI Z, ALAM Z, IANEVSKI A, et al. Interactive visual analysis of drug-target interaction networks using drug target profiler, with applications to precision medicine and drug repurposing[J]. Briefings in bioinformatics, 2020, 21(1): 211–220. [216] KIM S, CHEN Jie, CHENG Tiejun, et al. PubChem 2023 update[J]. Nucleic acids research, 2023, 51(D1): D1373–D1380. doi: 10.1093/nar/gkac956 [217] ZHENG Shuyu, ALDAHDOOH J, SHADBAHR T, et al. DrugComb update: a more comprehensive drug sensitivity data repository and analysis portal[J]. Nucleic acids research, 2021, 49(W1): W174–W184. doi: 10.1093/nar/gkab438 [218] LIU Hui, ZHANG Wenhao, ZOU Bo, et al. DrugCombDB: a comprehensive database of drug combinations toward the discovery of combinatorial therapy[J]. Nucleic acids research, 2020, 48(D1): D871–D881. [219] YANG W, SOARES J, GRENINGER P, et al. Genomics of drug sensitivity in cancer (GDSC): a resource for therapeutic biomarker discovery in cancer cells[J]. Nucleic acids res, 2013, 41(database issue): D955–D961. [220] AVRAM S, WILSON T B, CURPAN R, et al. DrugCentral 2023 extends human clinical data and integrates veterinary drugs[J]. Nucleic acids research, 2023, 51(D1): D1276–D1287. doi: 10.1093/nar/gkac1085 [221] MARTIN F J, AMODE M R, ANEIJA A, et al. Ensembl 2023[J]. Nucleic acids research, 2023, 51(D1): D933–D941. doi: 10.1093/nar/gkac958 [222] CONSORTIUM U. UniProt: the universal protein knowledgebase in 2023[J]. Nucleic acids research, 2023, 51(D1): D523–D531. doi: 10.1093/nar/gkac1052 [223] KANEHISA M, GOTO S. KEGG: Kyoto encyclopedia of genes and genomes[J]. Nucleic acids research, 2000, 28(1): 27–30. doi: 10.1093/nar/28.1.27 [224] OUGHTRED R, RUST J, CHANG C, et al. The BioGRID database: a comprehensive biomedical resource of curated protein, genetic, and chemical interactions[J]. Protein science: a publication of the protein society, 2021, 30(1): 187–200. doi: 10.1002/pro.3978 [225] NUSINOW D P, SZPYT J, GHANDI M, et al. Quantitative proteomics of the cancer cell line encyclopedia[J]. Cell, 2020, 180(2): 387–402.e16. doi: 10.1016/j.cell.2019.12.023 [226] LUNA A, ELLOUMI F, VARMA S, et al. CellMiner cross-database (CellMinerCDB) version 1.2: exploration of patient-derived cancer cell line pharmacogenomics[J]. Nucleic acids research, 2021, 49(D1): D1083–D1093. doi: 10.1093/nar/gkaa968 [227] CONSORTIUM W. Protein data bank: the single global archive for 3D macromolecular structure data[J]. Nucleic acids research, 2019, 47(D1): D520–D528. doi: 10.1093/nar/gky949 [228] SZKLARCZYK D, KIRSCH R, KOUTROULI M, et al. The STRING database in 2023: protein-protein association networks and functional enrichment analyses for any sequenced genome of interest[J]. Nucleic acids research, 2023, 51(D1): D638–D646. doi: 10.1093/nar/gkac1000 [229] KESHAVA PRASAD T S, GOEL R, KANDASAMY K, et al. Human protein reference database: 2009 update[J]. Nucleic acids research, 2009, 37(Database issue): D767–D772. [230] OCHOA D, HERCULES A, CARMONA M, et al. The next-generation open targets platform: reimagined, redesigned, rebuilt[J]. Nucleic acids research, 2023, 51(D1): D1353–D1359. doi: 10.1093/nar/gkac1046 [231] PIÑERO J, RAMÍREZ-ANGUITA J M, SAÜCH-PITARCH J, et al. The DisGeNET knowledge platform for disease genomics: 2019 update[J]. Nucleic acids research, 2020, 48(D1): D845–D855. [232] FORBES S A, BEARE D, BOUTSELAKIS H, et al. COSMIC: somatic cancer genetics at high-resolution[J]. Nucleic acids research, 2017, 45(D1): D777–D783. doi: 10.1093/nar/gkw1121 [233] KUHN M, LETUNIC I, JENSEN L J, et al. The SIDER database of drugs and side effects[J]. Nucleic acids research, 2016, 44(D1): D1075–D1079. doi: 10.1093/nar/gkv1075 [234] DAVIS A P, WIEGERS T C, JOHNSON R J, et al. Comparative toxicogenomics database (CTD): update 2023[J]. Nucleic acids research, 2023, 51(D1): D1257–D1262. doi: 10.1093/nar/gkac833 [235] DAVIS M I, HUNT J P, HERRGARD S, et al. Comprehensive analysis of kinase inhibitor selectivity[J]. Nature biotechnology, 2011, 29(11): 1046–1051. doi: 10.1038/nbt.1990 [236] TANG Jing, SZWAJDA A, SHAKYAWAR S, et al. Making sense of large-scale kinase inhibitor bioactivity data sets: a comparative and integrative analysis[J]. Journal of chemical information and modeling, 2014, 54(3): 735–743. doi: 10.1021/ci400709d [237] METZ J T, JOHNSON E F, SONI N B, et al. Navigating the kinome[J]. Nature chemical biology, 2011, 7(4): 200–202. doi: 10.1038/nchembio.530 [238] YAMANISHI Y, ARAKI M, GUTTERIDGE A, et al. Prediction of drug-target interaction networks from the integration of chemical and genomic spaces[J]. Bioinformatics, 2008, 24(13): i232–i240. doi: 10.1093/bioinformatics/btn162 [239] HUANG Kexin, FU Tianfan, GAO Wenhao, et al. Therapeutics data commons: machine learning datasets and tasks for drug discovery and development[EB/OL]. (2021–02–18)[2023–08–19]. http://arxiv.org/abs/2102.09548. [240] MIN Yaosen, WEI Ye, WANG Peizhuo, et al. Predicting the protein-ligand affinity from molecular dynamics trajectories[EB/OL]. (2022–08–19) [2023–08–19]. https://arxiv.org/abs/2208.10230.






























