
Prediction of multitype protein interactions combining Doc2vec and GCN
-
摘要: 多类型蛋白质−蛋白质相互作用(protein-protein interaction, PPI)的研究是从系统角度理解生物过程和揭示疾病机制的基础。现有的GNN-PPI、PIPR等针对多类型PPI预测方法在采用广度和深度优先搜索对数据集进行划分时,测试准确率会显著下降,因此本文基于Doc2vec方法思想和图卷积神经网络(graph convolutional network, GCN)技术,提出了一种新的多类型PPI预测方法GDP(GCN Doc2vec PPI )。该方法无需依赖蛋白质的物理和生物学特性,仅用序列信息对蛋白质进行编码,并结合网络结构信息对蛋白质进行特征聚合形成PPI信息,从而对其进行多类型预测。实验结果表明,该方法在不同规模的真实数据中可以有效地提高多类型PPI预测准确率,尤其是在训练集中未曾见过的新蛋白质之间的PPI。Abstract: The study of multitype protein-protein interactions (PPIs) is the basis for understanding biological processes and revealing disease mechanisms from a systematic perspective. Existing prediction methods for multiple types of PPIs, such as GNN-PPI and PIPR, show a considerable decline in test accuracy when the breadth- and depth-first searches are used to divide data sets. Therefore, this paper proposes a new multitype PPI prediction method (GDP) based on the Doc2vec method and graph convolutional neural network technology, which does not need to rely on the physical and biological properties of proteins. Moreover, the method only uses sequence information to encode proteins and combines the network structure information to conduct characteristic protein polymerization for developing PPI information to perform multitype prediction. Experimental results show that this method can effectively improve the prediction accuracy of multiple type PPIs in real data with different scales, especially in PPI between new proteins that have not been previously observed in the training set.
-
蛋白质−蛋白质相互作用(protein-protein interaction, PPI)在许多生物过程中都有着重要作用,在这些过程中,蛋白质通过与其他蛋白质相互作用形成特定功能。建立准确的 PPI 预测模型对于理解正常及疾病状态下的细胞生物至关重要,推动了现代医学的发展,如靶点治疗[1]和新药设计[2]。
生物实验技术[3-5]虽然能够直接发现和验证PPI,但价格昂贵、检测周期长,最显著的缺点是单个实验检测PPI会存在假阳性和假阴性的可能,因此其类型并不能得到完全解释[6-7]。随着高通量实验技术的迅速发展,PPI有关数据日益增多[8],这也使得通过计算方法预测其功能类型成为可能。相较生物实验技术,计算方法速度快、成本低,可以在短时间内预测一些高置信度的PPI。利用大量的PPI数据,可以构建蛋白质相互作用网络,进而通过复杂网络理论和机器学习方法预测PPI类型。其中,网络中的节点表示蛋白质,节点之间的连接表示对应蛋白质之间的相互作用。
针对PPI预测问题,国内外已有大量相关研究。文献[9]基于同源性的方法,通过计算蛋白质的BLAST值将一对序列映射到已知的相互作用蛋白质,从而推断出新的PPI;文献[10]基于相邻效应,提出结合自协方差(auto covariance, AC)和支持向量机(support vector machine, SVM)方法,利用氨基酸与其 30 个邻位氨基酸的相互作用表征 PPI 信息;文献[11]采用物理化学特性响应矩阵将序列转化为矩阵,使用局部相位量化的纹理描述符提取局部短语信息矩阵,将随机森林(random forest, RF)模型与新特征表示相结合来检测 PPI;文献[12]基于检测交互的实验技术,采用逻辑回归(logistic regression, LR)来预测交互类型;文献[13]基于SVM,结合描述氨基酸的联合三元组特征和序列信息来预测PPI。其中多类型PPI预测是对传统PPI预测方法的一种扩展和改进,需要提供更全面、准确和细致的预测结果。虽然基于计算方法和机器学习提出了用于多类型PPI预测的可行方法,但这些方法很大程度上依赖于提取和选择更好特征的能力,因此性能受到PPI特征表示和模型表达能力的限制。
近年来由于深度学习的发展,并在PPI预测问题上也得到了广泛应用。如文献[14-16]分别使用卷积神经网络(convolution neural network, CNN)、循环神经网络(recurrent neural network, RNN)以及区域卷积神经网络(region CNN, R-CNN)来提取序列中的高维信息特征,从而改进了PPI相关任务中的模型预测性能。相较于早期机器学习方法,以上模型有了一定的深度,非线性的建模能力得到了增强,对PPI预测这类复杂的任务表现也不断提升。
虽然上述方法能够高效地提取蛋白质序列信息,但忽视了PPI网络的结构信息,存在一定的局限性,准确性也有待提高。近年来,大量研究[17-18]表明,图神经网络在利用图结构信息方面有着显著的优势。因此,采用图神经网络(graph neural networks, GNN)以利用PPI网络的结构信息,搭建新型多类型PPI预测模型,对于提升预测的准确率有较好的前景。文献[19]考虑了PPI的相关性,提出使用GNN自动学习PPI网络中的蛋白质特征。文献[20]将GNN扩展到多类型PPI分类,并提出全新的测试集训练集划分方法以及“新蛋白质”这一概念——即在训练集中并没出现过的蛋白质,实验结果表明过往方法对“新蛋白质”的分类能力较弱。
因此,本文根据PPI网络中的蛋白质结点,利用其氨基酸序列信息和网络结构信息,对其进行多类型预测,提出一种融合Doc2vec[21]文本嵌入方法和图卷积网络(graph convolution network, GCN)[22]的多类型蛋白质相互作用分类预测模型。该模型利用自然语言处理领域中词袋预测任务的无监督模型,对蛋白质的氨基酸序列进行训练,并将模型的输出作为蛋白质序列信息的初步特征,随后使用一维卷积神经网络进行特征提取,并采用图神经网络作为下游模型,在对单个蛋白质进行表征的同时聚合它的邻居蛋白质的信息。该方法仅利用蛋白质序列信息和PPI网络结构信息,在有效处理任何长度的序列信息的同时也简化了模型深度,进而高效准确地预测蛋白质之间的相互作用,尤其是对于未曾见过的“新蛋白质”之间PPI的多类型预测。
1. 问题建模
假设存在一个蛋白质为点的集合P,蛋白质相互作用为边的集合V(即PPIs),相互作用的类型为标签的集合T,表达公式如下:
$$ \begin{gathered} P = \{ {p_1},{p_2},\cdots,{p_n}\} \\ V = \{ {v_{ij}} = \{ {p_i},{p_j}\} |{p_i},{p_j} \in P,I{\text{(}}{{{v}}_{{{ij}}}}{\text{)}} \in {\text{\{ 0,1\} }}\} \\ T = {\text{\{ }}{{{t}}_{\text{1}}}{\text{,}}{{{t}}_{\text{2}}}{\text{,}}\cdots{\text{,}}{{{t}}_{{m}}}{\text{\} }} \end{gathered} $$ (1) 式中: n表示蛋白质的个数;I表示相互作用,当I(vij) =1/0时,表明蛋白质pi和pj间存在/不存在相互作用(或它们之间的相互作用尚未发现);m表示在数据集中出现的相互作用的类别总个数。
对于每一条蛋白质相互作用vij,设其标签为xij,且xij∈T。所有的蛋白质相互作用集合和对应的标签集合构成了所需的数据集的集合D,所有的蛋白质相互作用集合和蛋白质集合构成了蛋白质互作网络G,表达公式如下:
$$ \begin{gathered} D = \{ ({v_{ij}},{x_{ij}})|{v_{ij}} \in V,{x_{ij}} \in T\} \\ G = \{ P,V\} \end{gathered} $$ (2) 由上述可知,针对多类型PPI分类预测任务,需要构建一个模型,并在数据集D中划分训练集和测试集,从训练集中学习使得该模型预测出的
$ {\hat x_{ij}} $ 不断地接近于真实值xij。2. GDP预测模型
2.1 预测方法
本文提出融合Doc2vec与GCN的多类型蛋白质相互作用预测方法,该方法主要分为蛋白质嵌入模块、特征提取模块、图卷积编码模块和分类器预测模块4个部分。蛋白质嵌入模块通过调整Doc2vec非监督段落向量学习模型,将不定长的蛋白质序列特征信息嵌入至低维向量空间,解决了蛋白质初步特征选取问题;特征提取模块利用一维卷积网络的堆叠,将蛋白质嵌入模块获得的特征进一步整合,利用多个卷积核,放大针对PPI多分类预测的有效特征信息;图卷积编码模块利用图深度学习的优势,充分结合PPI网络结构的信息,聚合每个蛋白质的相邻蛋白质的信息,优化了蛋白质结点的编码表征问题;分类器预测模块根据PPI网络结构信息,找到蛋白质相互作用边,结合两个蛋白质节点信息,并不断从中学习更高效且准确的分类预测;具体结构如图1所示。
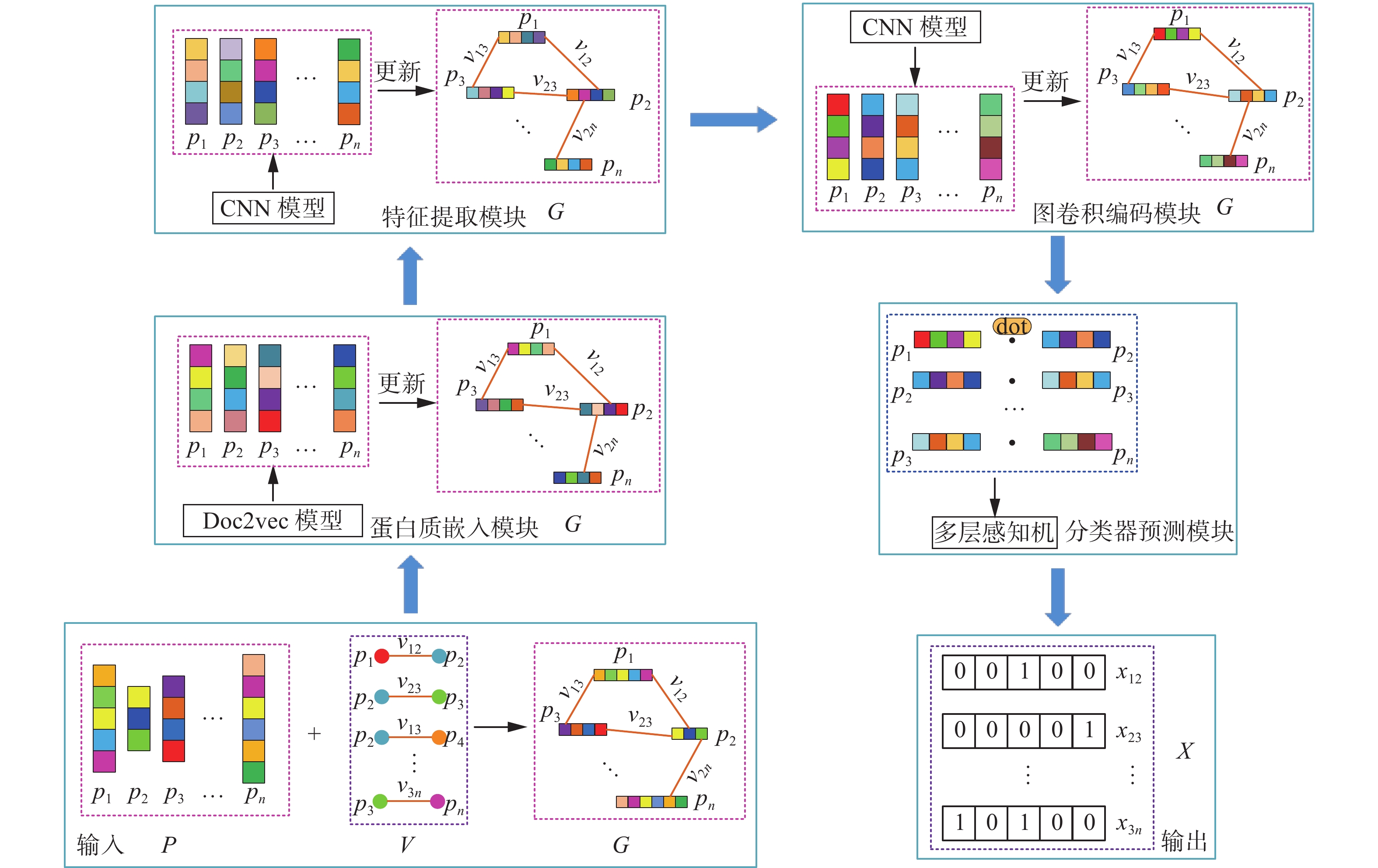 图 1 GDP框架结构Fig. 1 GDP framework
图 1 GDP框架结构Fig. 1 GDP framework 下载:
全尺寸图片
下载:
全尺寸图片
2.2 蛋白质嵌入模块
蛋白质序列嵌入一直是生物信息学领域的重要问题,良好的表征能力决定了蛋白质预测相关任务的上限。随着Word2vec、Seq2vec等自然语言处理(natural language processing, NLP)领域中词句嵌入技术发展,凭借其强大的表征能力,近年来已被应用于蛋白质的相关表征任务中。Doc2vec是其中嵌入方法的一种,能得到任意长度文档的向量表示。基于此,本文将蛋白质序列看作文档,以改进Doc2vec方法对蛋白质序列进行嵌入,模块结构如图2所示。由图2可知,本文将每个蛋白质p的氨基酸序列s作为输入,设置超参数滑动窗口长度w和子序列数量k,其中每个子序列由若干个k-mer(k个氨基酸可以组合为一个k-mer)构成。对于每个子序列采取连续词袋(continuous bag of words)模型训练,即使用子序列的嵌入和滑动窗口中的上下文k-mer的嵌入来学习预测中央k-mer出现的概率。聚合k个子序列的嵌入信息得到当前输入蛋白质的序列嵌入。通过该模块可以将最终生成的低维向量作为多标签分类任务的初步特征。
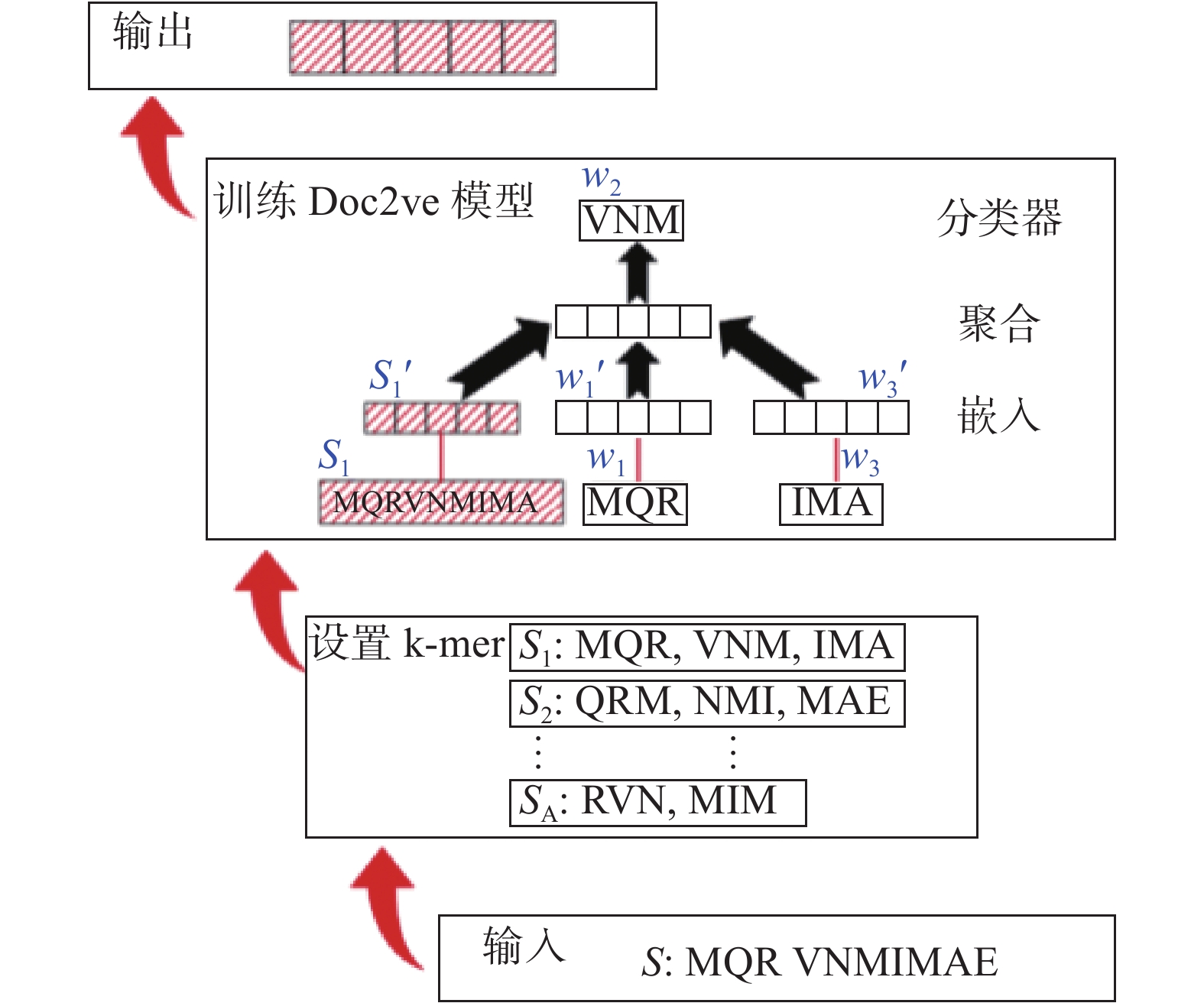 图 2 蛋白质嵌入模块结构Fig. 2 Protein embedding framework下载:
全尺寸图片
图 2 蛋白质嵌入模块结构Fig. 2 Protein embedding framework下载:
全尺寸图片
2.3 特征提取模块
在针对NLP中文本任务等序列任务时,一维卷积神经网络有着提升网络特征表达、高效升维与降维、跨通道信息交互等优点,故本文采用了一维卷积神经网络来更深层地提取蛋白质的局部特征信息,该模块将蛋白质嵌入模块得到的特征作为输入,经过卷积与全连接层作为输出,公式如下:
$$ {\boldsymbol{h}}_k^v = f\left(\sum\limits_{i = 1}^N {{\boldsymbol{h}}_i^{v - 1}*{\boldsymbol{w}}_k^v + {\boldsymbol{b}}_k^v} \right) $$ (3) 式中:
$ {\boldsymbol{h}}_k^v $ 为蛋白质v层第k次卷积映射,f为激活函数,N为卷积的数量,*为卷积操作,$ {\boldsymbol{w}}_k^v $ 为权值,$ {\boldsymbol{b}}_k^v $ 为偏置量。其中本文采用Relu激活函数防止梯度消失,采用最大值池化操作提取主要特征。经过两层的卷积再连接一层全连接层,该模块能够全面观测蛋白质序列信息并提取到针对多类型PPI预测任务的有效特征,提高模型的分类效率。
2.4 图卷积编码模块
GNN是基于深度学习的处理图域信息的方法,由于其较好的性能和可解释性,GNN 已成为一种广泛应用的图分析方法[23];生物计算主要利用了蛋白质相互作用网络,因此基于GNN进行相关生物任务取得了高效的进展。GNN是对图进行特征变换和特征提取,需要尽可能多的利用图中节点特征和拓扑信息。图分类相关任务中,目前主要有两种卷积方式:1) 信息传递式的卷积,即直接在原始图结构中定义由邻居聚合和迭代更新机制所组成的卷积算子,例如GCN、图注意力网络(graph attention network, GAT)等;2) 传统CNN式的卷积,先将非欧氏图转化为规则网格结构,再应用传统卷积神经网络直接进行卷积操作。
图同构卷积网络(GINConv)属于GCN中的一种,其在同构网络上有强大的表征能力,故本文采用GINConv,图卷积编码结构如图3所示。
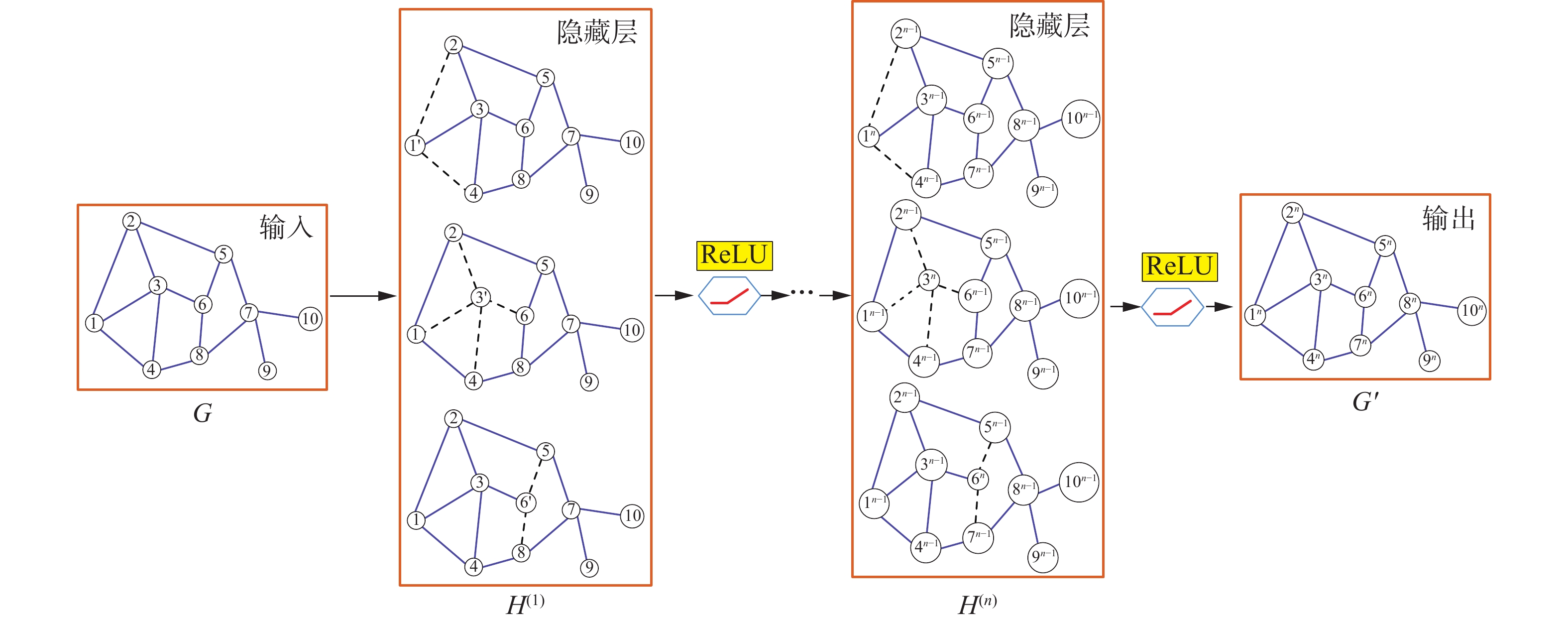 图 3 图卷积编码结构Fig. 3 Graph convolution encoding framework下载:
全尺寸图片
图 3 图卷积编码结构Fig. 3 Graph convolution encoding framework下载:
全尺寸图片
GINConv将卷积过程形式化为信息传递和节点信息更新两个函数,各个节点将自己邻居的信息聚合到自身节点,节点信息更新是将该节点上一层的节点表示与聚合后的邻居信息进行结合,具体过程如下公式:
$$ {\boldsymbol{h}}_v^k = \varphi ({\boldsymbol{h}}_v^{k - 1},f(\{ {\boldsymbol{h}}_u^{k - 1}:u \in N(v)\} )) $$ (4) 式中:
${\boldsymbol{h}}_v^k$ 是蛋白质$ {p_v} $ 在第k层的向量表示,$\varphi$ 表示映射函数,N(v)表示$ {p_v} $ 的邻居节点集合,f是处理邻居节点的函数。本文采用多层感知机(multilayer perceptrons, MLP)作为映射函数,累加作为邻居的信息聚合,则上述更新函数f可表示为$$ {\boldsymbol{h}}_v^k = {\text{ML}}{{\text{P}}^{{k}}}\left((1 + {\varepsilon ^k}) \cdot {\boldsymbol{h}}_v^{{{{\rm{k}} - 1}}} + \sum\limits_{u \in {\text{N}}(v)} {{\boldsymbol{h}}_u^{k - 1}} \right) $$ (5) 其中
$ \varepsilon $ 可以是超参数或者为可学习参数。2.5 分类器模块
通过以上3个模块,每个蛋白质都学习到了自身的表征向量,利用点积运算将蛋白质pi和pj的表征向量结合起来,在后续添加一层MLP作为分类器,来进行多类型PPI预测。预测的结果表示为
${\hat x_{ij}} = {{\rm{MLP}}} ({{\boldsymbol{h}}_i} \cdot {{\boldsymbol{h}}_j})$ ,其中hi和hj为图卷积编码模块对应蛋白质的输出。2.6 损失函数
对于该任务,本文采用多任务二元交叉熵作为损失函数,公式如下:
$$ L_{\text{loss}} = - \sum\limits_{k = 1}^n {\left(\sum\limits_{{v_{ij}} \in {V_{{\text{train}}}}} {(1 - x_{ij}^k){\text{log(}}1 - y_{ij}^k) + x_{ij}^k{\text{log}}y_{ij}^k} \right)} $$ (6) 式中:Vtrain表示PPI集合V中划分出的训练集,
$ x_{ij}^k $ 表示训练集中vij对应的第k种功能类型的真实标签,$ \hat x_{ij}^k $ 则表示模型对其预测的输出。3. 实验结果与分析
3.1 实验数据和评价指标
本文使用String数据库[24]中的多类型PPI数据作为其中一个数据集来评估所提出GDP预测模型,String数据库收集整合了公开的蛋白质相互作用信息来源,并构建了一个全面客观的大型PPI网络,包括直接(物理)和间接(功能)相互作用,其将PPI分为7种类型,即反应(reaction)、结合(binding)、(activation)、抑制(inhibition)、催化(catalysis)和表达(expression),任意一对PPI至少包含其中一种类型。此外,为验证GDP模型的泛用性,运用了Chen等[16]从智人子集中随机生成的SHS27k和SHS148k两个子数据集。3个数据集的信息如表1所示。
表 1 数据集的规模信息Table 1 The size of the data set数据集 蛋白质数量 PPI个数 String 15335 593397 SHS27k 1690 7624 SHS148k 5189 44488 为避免数据的极度不平衡对结果造成不良影响,采用
$F_{1,{\rm{micro}}}$ 得分作为评价指标。公式如下:$$ F_{1,{\text{micro}}} = 2\frac{{{{R_{{\rm{recall}}}}} \times {{{P_{{\rm{precision}}}}}}}}{{{{R_{{\rm{recall}}}}} + {{{P_{{\rm{precision}}}}}}}} $$ $$ {{{{R_{{\rm{recall}}}}}}} = \frac{{\displaystyle\sum\limits_{i = 1}^n {{T_{P,i}}} }}{{\displaystyle\sum\limits_{i = 1}^n {{T_{P,i}}} + \displaystyle\sum\limits_{i = 1}^n {{F_{N,i}}} }} $$ (7) $$ {{{{P_{{\rm{precision}}}}}}} = \frac{{\displaystyle\sum\limits_{i = 1}^n {{T_{P,i}}} }}{{\displaystyle\sum\limits_{i = 1}^n {{T_{P,i}}} + \displaystyle\sum\limits_{i = 1}^n {{F_{P,i}}} }} $$ 式中:n为分类类别总数;TP,i表示第i类的真阳性数;FP,i表示第i类的假阳性数;FN,i表示第i类的假阴性数。
3.2 实验设置
本文实验运行环境为Win10系统、32 GB内存,利用Pycharm软件和Pytorch1.8版本框架搭建GDP预测模型。实验的参数设置如表2所示。
表 2 实验参数Table 2 Experimental parameter类型 超参数 数值 模型结构参数 k-mer 3 图卷积层数 2 蛋白质嵌入维度 128 特征提取维度 128 图卷积嵌入维度 128 优化方法 Adam 模型训练参数 学习率 0.001 批处理大小 1024 迭代次数 300 3.3 实验结果
为验证本文提出的方法的有效性,对上述3个数据集分别使用随机(Random)搜索、广度优先搜索(breadth first search, BFS)和深度优先搜索(depth first search, DFS)策略进行划分。如图4所示,当分别使用3种策略对数据集进行划分时,在选取相同数量的PPI情况下,BFS和DFS划分策略下的测试集蛋白质节点远少于Random划分策略,即采用BFS和DFS划分数据集时,能够出现大量训练集未出现过的“新蛋白质”,这些新蛋白质更能检测模型的预测效率。因此,本文在上述3个数据集采用Random、BFS和DFS 3种划分方式,分别与当前的PPI分类方法[11,12,14-16,20]进行了对比实验,其中RF与LR分别使用随机森林和逻辑回归方法,DPPI、DNN-PPI和PIPR使用卷积网络方法,GNN-PPI采用图神经网络方法。实验结果分别如图5和图6所示。
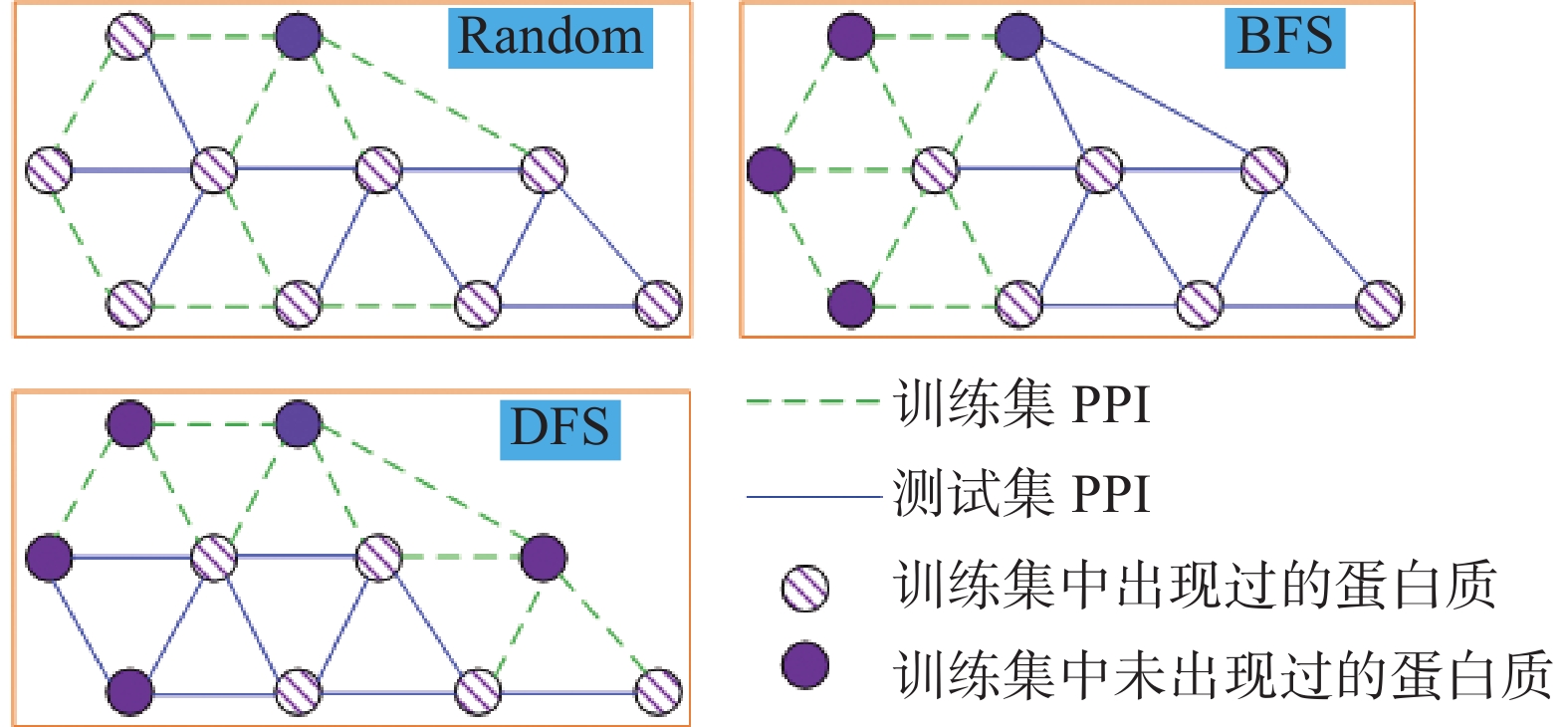 图 4 不同的测试集划分策略Fig. 4 Different test sets partitioning strategies下载:
全尺寸图片
图 4 不同的测试集划分策略Fig. 4 Different test sets partitioning strategies下载:
全尺寸图片
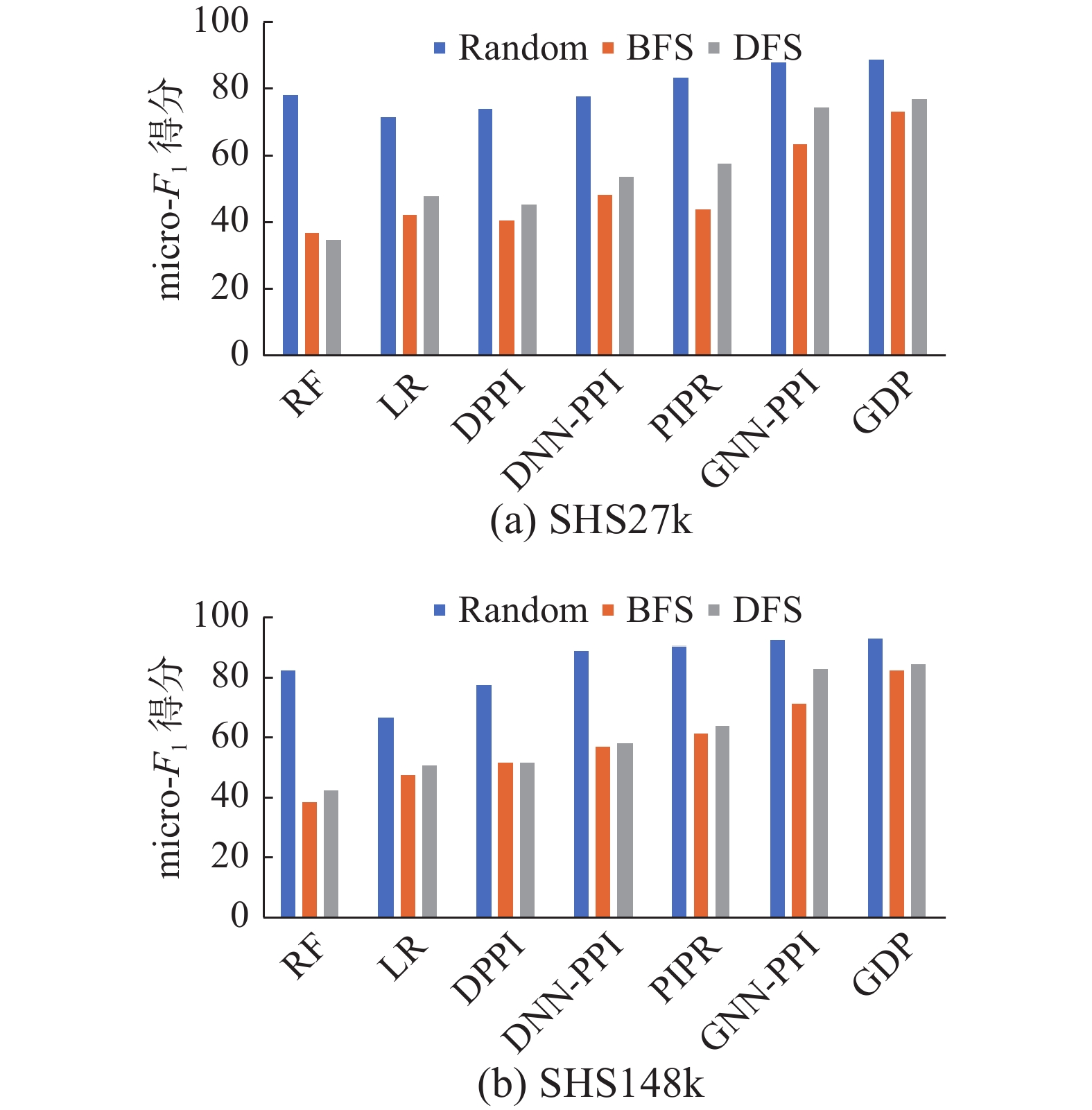 图 5 各方法在数据集SHS27k和SHS148k上的micro-F1得分Fig. 5 Micro-F1 score of each method on SHS27k and SHS148k dataset下载:
全尺寸图片
图 5 各方法在数据集SHS27k和SHS148k上的micro-F1得分Fig. 5 Micro-F1 score of each method on SHS27k and SHS148k dataset下载:
全尺寸图片
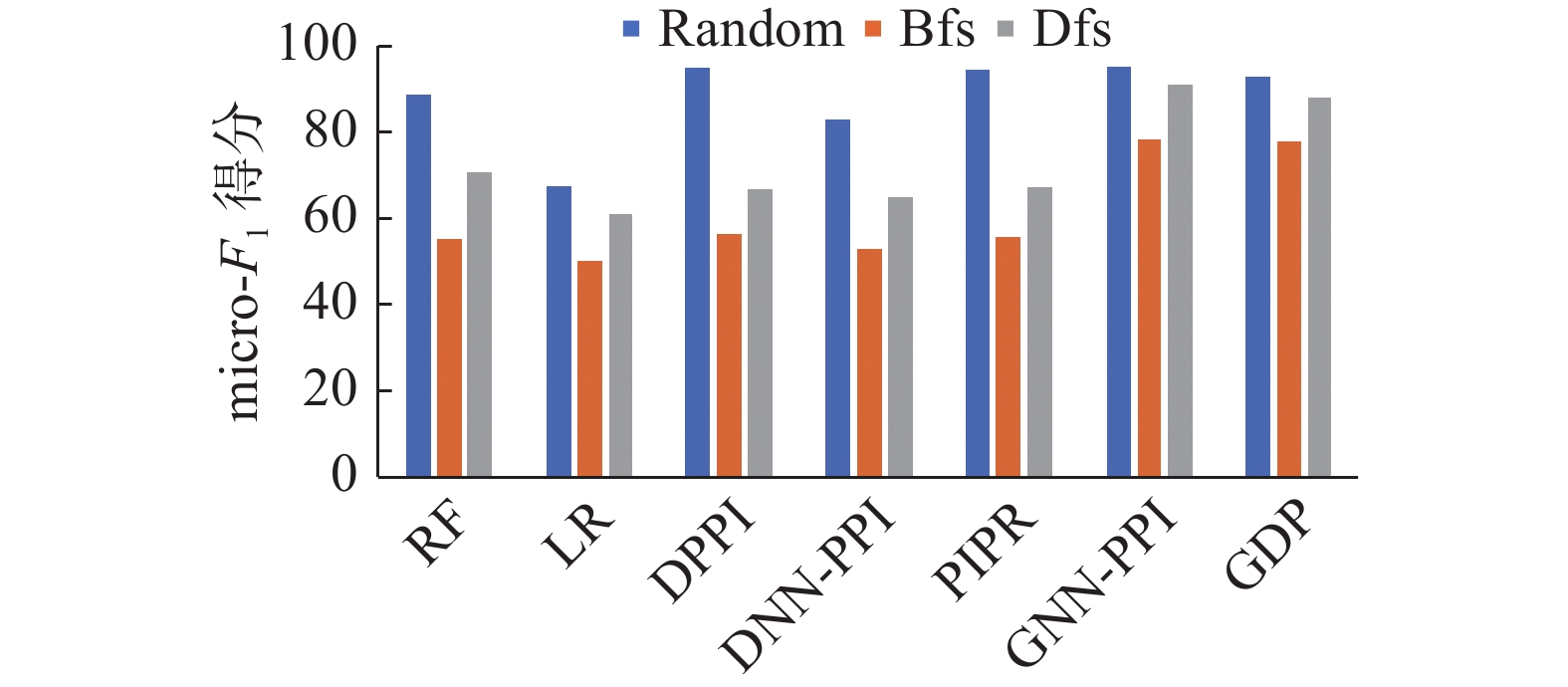 图 6 各方法在数据集String上的micro-F1得分Fig. 6 Micro-F1 score of each method on String dataset下载:
全尺寸图片
图 6 各方法在数据集String上的micro-F1得分Fig. 6 Micro-F1 score of each method on String dataset下载:
全尺寸图片
由图5可知,在micro-F1得分指标和3种数据集划分模式下,本文提出的GDP方法在SHS27k数据集和SHS148k数据集上的效果均优于其他方法。在数据集SHS27k中,GDP方法在Random、BFS和DFS等3种划分方式下的micro-F1得分指标相较于目前性能最好的GNN-PPI方法分别提升了1.2%、9.1%和3.5%;在数据集SHS148k中,GDP方法在Random、BFS和DFS等3种划分方式下的micro-F1指标分别提升了0.8%、11.4%和1.9%。由此可知,GDP方法的多类型PPI预测结果的准确率取得了较大的提升,其原因是PPI网络中仅缺失部分边缘蛋白质,而对蛋白质进行特征表示时能够获得大部分邻居的特征表示。实验结果也表明,使用图卷积能够较好的聚合邻居节点特征的效果,能够较大提升图网络中的预测任务结果。与此同时,在Random和DFS模式下,GDP方法也取得了一定提升,这表明蛋白质序列表征在PPI任务中有着举足轻重的作用[25]。
由图6可知,在micro-F1得分指标和3种数据集划分模式下,GDP方法在String数据集上的效果优于大部分算法。但在BFS和DFS划分策略下,GDP方法略逊色于GNN-PPI方法,而在Random划分策略下,传统氨基酸特征提取的深度学习方法DPPI和PIPR也稍高于GDP方法。其原因是String数据集属于大规模PPI网络,而GDP方法训练参数小,网络深度浅,对于大型网络易出现过拟合的现象,这也表明GDP方法存在一定的局限性。
为进一步验证GDP方法中设计的蛋白质嵌入模块,特征提取模块,图卷积编码模块的有效性,以及这3个模块对于整个方法性能的提升,本文将GDP方法转化为3个新的方法:GDP-ACID、GDP-CNN与GDP-GNN方法。GDP-ACID将蛋白质编码模块替换为传统氨基酸One-hot编码方式,GDP-CNN将特征提取模块替换为两层MLP的堆叠,GDP-GNN则删除了图卷积编码模块。实验结果如表3所示,由表3可知,当替换或删减了这3个模块后,在不同数据集上和不同划分策略下,预测效果都会出现一定程度的下滑。相较于蛋白质嵌入模块,图卷积模块对整个方法的影响更为明显,这也反映了将图深度学习应用到PPI网络上的必要性。为研究不同蛋白质嵌入维度与图卷积嵌入维度对micro-F1指标的影响,在中等规模数据集SHS148K上分别设置不同的蛋白质嵌入维度d1与图卷积嵌入维度d2,实验结果如表4和5所示。由表4和5可知,随着嵌入维度的不断增加,micro-F1指标得分略微降低,但由于增大嵌入维度,可将更多的信息编码,故其收敛速度加快,较好地提升了方法的性能。另一方面,嵌入的维度过高时会造成过拟合的现象。因此为选择合适的嵌入维度,本文将蛋白质嵌入维度d1与图卷积嵌入维度d2都设置为128。
表 3 GDP方法及其相关方法在不同数据集和划分策略上的micro-F1得分Table 3 Micro-F1 scores of the GDP method and its relative on different data sets and partitioning strategies数据集 划分
策略GDP GDP-ACID GDP-CNN GDP-GNN SHS27K Random 88.67 83.87 80.21 72.76 BFS 72.68 60.98 56.25 58.32 DFS 77.23 58.75 62.87 52.67 SHS148K Random 92.56 90.93 88.53 75.36 BFS 82.21 73.54 69.82 63.21 DFS 84.17 72.46 76.23 70.87 String Random 91.24 85.78 80.61 81.33 BFS 77.91 69.67 73.54 70.65 DFS 84.93 80.23 79.98 72.93 表 4 不同嵌入维度d1对GDP方法micro-F1指标的影响Table 4 Effects of different embedding dimensions d1 on micro-F1 index of GDP method划分策略 d1 32 64 128 256 Random 89.34 90.27 92.56 91.03 BFS 75.48 80.36 82.21 79.93 DFS 82.36 84.53 84.17 80.48 表 5 不同嵌入维度d2对GDP方法micro-F1指标的影响Table 5 Effects of different embedding dimensions d2 on micro-F1 index of GDP method划分策略 d2 32 64 128 256 Random 88.52 91.98 92.56 92.79 BFS 73.72 81.22 82.21 78.66 DFS 83.39 82.74 84.17 82.91 4. 结束语
针对多类型蛋白质相互作用预测问题,本文提出一种融合Doc2vec与GCN的预测方法,GDP方法改进了Doc2vec方法,在不依赖于生物特性信息的情况下,充分地利用了其完整氨基酸序列信息,为下游模型的输入提供了有效的特征,同时将图深度学习运用到PPI网络中,通过图卷积聚合邻居蛋白质的特征信息,考虑了整个网络的结构信息。在真实数据集上与多种其它类似算法进行对比,实验结果表明本文提出的GDP预测模型具有更高的准确性。
后续工作中,将从两个角度进一步研究:一是选择更高效的模型对蛋白质序列进行嵌入表征,如基于Transformer方法,该方法能将蛋白质的GO注释以及二级结构结合起来表征蛋白质,信息利用全面并且能够看见全局的序列特征;二是探究图深度学习领域对蛋白质相互作用网络其他相关任务的影响,如蛋白质的结构预测或者PPI网络比对任务。
-
图 1 GDP框架结构
Fig. 1 GDP framework
下载:
全尺寸图片
图 2 蛋白质嵌入模块结构
Fig. 2 Protein embedding framework
下载:
全尺寸图片
图 3 图卷积编码结构
Fig. 3 Graph convolution encoding framework
下载:
全尺寸图片
图 4 不同的测试集划分策略
Fig. 4 Different test sets partitioning strategies
下载:
全尺寸图片
图 5 各方法在数据集SHS27k和SHS148k上的micro-F1得分
Fig. 5 Micro-F1 score of each method on SHS27k and SHS148k dataset
下载:
全尺寸图片
图 6 各方法在数据集String上的micro-F1得分
Fig. 6 Micro-F1 score of each method on String dataset
下载:
全尺寸图片
表 1 数据集的规模信息
Table 1 The size of the data set
数据集 蛋白质数量 PPI个数 String 15335 593397 SHS27k 1690 7624 SHS148k 5189 44488 表 2 实验参数
Table 2 Experimental parameter
类型 超参数 数值 模型结构参数 k-mer 3 图卷积层数 2 蛋白质嵌入维度 128 特征提取维度 128 图卷积嵌入维度 128 优化方法 Adam 模型训练参数 学习率 0.001 批处理大小 1024 迭代次数 300 表 3 GDP方法及其相关方法在不同数据集和划分策略上的micro-F1得分
Table 3 Micro-F1 scores of the GDP method and its relative on different data sets and partitioning strategies
数据集 划分
策略GDP GDP-ACID GDP-CNN GDP-GNN SHS27K Random 88.67 83.87 80.21 72.76 BFS 72.68 60.98 56.25 58.32 DFS 77.23 58.75 62.87 52.67 SHS148K Random 92.56 90.93 88.53 75.36 BFS 82.21 73.54 69.82 63.21 DFS 84.17 72.46 76.23 70.87 String Random 91.24 85.78 80.61 81.33 BFS 77.91 69.67 73.54 70.65 DFS 84.93 80.23 79.98 72.93 表 4 不同嵌入维度d1对GDP方法micro-F1指标的影响
Table 4 Effects of different embedding dimensions d1 on micro-F1 index of GDP method
划分策略 d1 32 64 128 256 Random 89.34 90.27 92.56 91.03 BFS 75.48 80.36 82.21 79.93 DFS 82.36 84.53 84.17 80.48 表 5 不同嵌入维度d2对GDP方法micro-F1指标的影响
Table 5 Effects of different embedding dimensions d2 on micro-F1 index of GDP method
划分策略 d2 32 64 128 256 Random 88.52 91.98 92.56 92.79 BFS 73.72 81.22 82.21 78.66 DFS 83.39 82.74 84.17 82.91 -
[1] PETTA I, LIEVENS S, LIBERT C, et al. Modulation of protein-protein interactions for the development of novel therapeutics[J]. Molecular therapy, 2016, 24(4): 707–718. doi: 10.1038/mt.2015.214 [2] SKRABANEK L, SAINI H K, BADER G D, et al. Computational prediction of protein-protein interactions[J]. Molecular biotechnology, 2008, 38(1): 1–17. doi: 10.1007/s12033-007-0069-2 [3] FIELDS S, SONG O K. A novel genetic system to detect protein-protein interactions[J]. Nature, 1989, 340(6230): 245–246. doi: 10.1038/340245a0 [4] GAVIN A C, BÖSCHE M, KRAUSE R, et al. Functional organization of the yeast proteome by systematic analysis of protein complexes[J]. Nature, 2002, 415(6868): 141–147. doi: 10.1038/415141a [5] HO Y, GRUHLER A, HEILBUT A, et al. Systematic identification of protein complexes in Saccharomyces cerevisiae by mass spectrometry[J]. Nature, 2002, 415(6868): 180–183. doi: 10.1038/415180a [6] SUN Tanlin, ZHOU Bo, LAI Luhua, et al. Sequence-based prediction of protein protein interaction using a deep-learning algorithm[J]. BMC bioinformatics, 2017, 18(1): 1–8. doi: 10.1186/s12859-016-1414-x [7] LECUN Y, BENGIO Y, HINTON G. Deep learning[J]. Nature, 2015, 521(7553): 436–444. doi: 10.1038/nature14539 [8] CIERPICKI T, GREMBECKA J. Targeting protein-protein interactions in hematologic malignancies: still a challenge or a great opportunity for future therapies?[J]. Immunological reviews, 2015, 263(1): 279–301. doi: 10.1111/imr.12244 [9] PHILIPP O, OSIEWACZ H D, KOCH I. Path2PPI: an R package to predict protein-protein interaction networks for a set of proteins[J]. Bioinformatics, 2016, 32(9): 1427–1429. doi: 10.1093/bioinformatics/btv765 [10] GUO Yanzhi, YU Lezheng, WEN Zhining, et al. Using support vector machine combined with auto covariance to predict protein-protein interactions from protein sequences[J]. Nucleic acids research, 2008, 36(9): 3025–3030. doi: 10.1093/nar/gkn159 [11] WONG L, YOU Zhuhong, LI Shuai, et al. Detection of protein-protein interactions from amino acid sequences using a rotation forest model with a novel PR-LPQ descriptor[C]//International Conference on Intelligent Computing. Cham: Springer, 2015: 713−720. [12] SILBERBERG Y, KUPIEC M, SHARAN R. A method for predicting protein-protein interaction types[J]. PLoS One, 2014, 9(3): e90904. doi: 10.1371/journal.pone.0090904 [13] SHEN Juwen, ZHANG Jian, LUO Xiaomin, et al. Predicting protein-protein interactions based only on sequences information[J]. Proceedings of the national academy of sciences of the United States of America, 2007, 104(11): 4337–4341. [14] LI Hang, GONG Xiujun, YU Hua, et al. Deep neural network based predictions of protein interactions using primary sequences[J]. Molecules, 2018, 23(8): 1923. doi: 10.3390/molecules23081923 [15] HASHEMIFAR S, NEYSHABUR B, KHAN A A, et al. Predicting protein-protein interactions through sequence-based deep learning[J]. Bioinformatics, 2018, 34(17): i802–i810. doi: 10.1093/bioinformatics/bty573 [16] CHEN Muhao, JU C J T, ZHOU Guangyu, et al. Multifaceted protein-protein interaction prediction based on siamese residual RCNN[J]. Bioinformatics, 2019, 35(14): i305–i314. doi: 10.1093/bioinformatics/btz328 [17] 吴国栋, 查志康, 涂立静, 等. 图神经网络推荐研究进展[J]. 智能系统学报, 2020, 15(1): 14–24. doi: 10.11992/tis.201908034 WU Guodong, ZHA Zhikang, TU Lijing, et al. Research advances in graph neural network recommendation[J]. CAAI transactions on intelligent systems, 2020, 15(1): 14–24. doi: 10.11992/tis.201908034 [18] 马帅, 刘建伟, 左信. 图神经网络综述[J]. 计算机研究与发展, 2022, 59(1): 47–80. doi: 10.7544/issn1000-1239.20201055 MA Shuai, LIU Jianwei, ZUO Xin. Survey on graph neural network[J]. Journal of computer research and development, 2022, 59(1): 47–80. doi: 10.7544/issn1000-1239.20201055 [19] YANG Fang, FAN Kunjie, SONG Dandan, et al. Graph-based prediction of Protein-protein interactions with attributed signed graph embedding[J]. BMC bioinformatics, 2020, 21(1): 323. doi: 10.1186/s12859-020-03646-8 [20] LYU Guofeng, HU Zhiqiang, BI Yanguang, et al. Learning unknown from correlations: graph neural network for inter-novel-protein interaction prediction[EB/OL]. (2021−05−14)[2022−12−30].https://arxiv.org/abs/2105.06709. [21] LE Q V, MIKOLOV T. Distributed representations of sentences and documents[EB/OL]. (2014−05−16) [2022−12−30].https://arxiv.org/abs/1405.4053. [22] ZHANG Si, TONG Hanghang, XU Jiejun, et al. Graph convolutional networks: a comprehensive review[J]. Computational social networks, 2019, 6(1): 1–23. doi: 10.1186/s40649-019-0061-6 [23] 万莹莹. 基于图卷积网络的半监督图分类研究[D]. 桂林: 广西师范大学, 2021. WAN Yingying. Research on semi-supervised graph classification with graph convolutional network[D]. Guilin: Guangxi Normal University, 2021. [24] SZKLARCZYK D, GABLE A L, LYON D, et al. STRING v11: protein-protein association networks with increased coverage, supporting functional discovery in genome-wide experimental datasets[J]. Nucleic acids research, 2019, 47(D1): D607–D613. doi: 10.1093/nar/gky1131 [25] 桂元苗. 面向蛋白互作预测的序列数据特征识别研究[D]. 合肥: 中国科学技术大学, 2019. GUI Yuanmiao. Research on feature recognition of sequence data for protein interaction prediction[D]. Hefei: University of Science and Technology of China, 2019.






















