

DOI: 10.11992/tis.201603034

 , , , , ,
, , , , ,
, , , , ,
, , , , ,
蛋白质分子是通过与其他蛋白质分子相互作用发挥功能的,近年来随着高通量技术的快速发展,海量的蛋白质相互作用数据被挖掘出来,从而形成蛋白质相互作用网络(protein-protein interaction, PPI)。网络医学近年来在计算医学领域发展迅速,PPI网络中的蛋白模块往往具有特定的生物功能。Barabasi等认为疾病的产生是由于PPI中某个局部的蛋白链接关系发生了紊乱[1],并进一步提出了拓扑模块、功能模块和疾病模块是存在相同的共有蛋白成员的。大家普遍认为在拓扑结构上链接比较紧密的蛋白在生物功能上也更加相似。基于这个假设,为了可以精确地寻找到与疾病相关的蛋白模块,需要先从PPI网络中检测出具有比较显著生物意义的功能模块。
目前功能模块的检测方法主要是使用复杂网络领域中的社团划分方法将PPI网络划分为多个拓扑模块,然后对这些拓扑模块再进行生物功能的检测。Bader等提出了一种叫做MCODE的方法,该方法首先根据节点的邻居对每一个节点赋一个权重,然后选择权重较大的节点作为种子节点进行社团划分[2]。该方法可以发现重叠的蛋白质功能模块。DPClus等使用类似的方法对网络中的每条边赋权重,然后选择权重最大的边的节点作为初始种子节点进行社团划分[3]。Edward等提出了一种基于熵的方法进行功能模块的检测,该方法首先随机选择一个节点作为种子节点,然后将该种子节点和其周围的邻居作为一个种子类,通过熵的减少来移除边界点和增加新节点形成蛋白模块,直到遍历完网络中的所有节点[4]。
上述功能模块划分算法主要是根据PPI中的链接关系,也就是只找到了在拓扑结构上链接紧密的模块。由于目前人类所获取的蛋白相互作用数据只获取了实际相互作用的10%~20%[5],所以PPI网络是比较稀疏的,使用传统的复杂网络中的社团划分方法并不能保证精确地找到具有某种生物功能的模块。蛋白质复合体(protein complex)是2个及其以上的蛋白相互作用而形成的复合物,一般分为结构型的蛋白质复合体和功能型蛋白质复合体2大类。目前关于蛋白质复合体的数据已经可以方便地获取,因此可以考虑将蛋白质复合体的数据融合到PPI网络中,从而可以提高功能模块的发现精度。
本文首先将蛋白质复合体数据融合到PPI网络中,然后使用K均值(K-Means)和非负矩阵分解(non-negative matrix factorization, NMF)2种算法对融合后的数据进行模块划分,针对得到的模块进行基因本体(gene ontology, GO)和通路(pathway)富集分析并进一步计算模块的GO同质性。
1 社团划分及模块生物学分析 1.1 PPI网络的表示PPI网络可以表示为一个无向无权图,其中V表示顶点集、E表示边集。矩阵A表示邻接矩阵,A的定义为
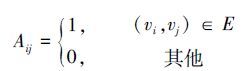 |
(1) |
式中:Aij表示节点i和节点j有连边,vi和vj表示节点i和节点j。
1.2 模块检测算法模块目前还没有一个统一的定义,大家对模块的共识是:模块内部的边比较紧密而模块之间的边要尽量稀疏[6]。本文主要使用K均值和非负矩阵分解2种算法对PPI网络进行模块检测。
1) K均值[7]
K均值是一个比较经典的聚类算法。给定一个含有N个节点的数据集x1, x2, …, xn,其中每个节点的维度是D维,将该数据集划分为k个类。每一类的类中心表示为μk,为每一个节点定义一个指示向量rnk,其物理含义是如果节点n的类标号为k,则值为1;否则为0。
K均值算法的主要思想就是所有样本点到各自的类中心的距离最短,其目标函数为
 |
(2) |
根据式(2) 可以得到类中心的迭代公式为
 |
(3) |
其代表的物理含义是第k个类中所有样本点的均值作为该类的类中心,然后其他节点根据与该类中心的距离来判断是不是属于这个类。通过不停地迭代,直到所有的类中心不在改变为止。
2) 非负矩阵分解
非负矩阵分解最早是由Lee 和Seung[8]提出的。若一个矩阵其所有的元素没有负数,这样的矩阵叫做非负矩阵。对一个n×m的非负矩阵X , 其行向量代表特征,列向量代表样本。非负矩阵分解的任务就是把X分解为两个非负矩阵使得X≈FGT,其中F是一个n×k的矩阵,G是m×k的矩阵(k为类的个数)。其目标函数为
 |
(4) |
式中:G为最后的划分矩阵。F和G的迭代规则如下:
 |
(5) |
当误差小于某个阈值a或达到最大迭代次数时算法终止,F矩阵描述了网络中节点隶属于某个社区的概率。
1.3 模块的富集分析为了确定每一个模块具体的生物功能,对每个模块分别进行GO和Pathway富集分析。每个模块会对所有的GO术语或者Pathway进行分析,并且返回具有最小P-value的GO术语或Pathway表示模块中的蛋白质在该GO术语或者Pathway中出现了富集,即该GO术语描述了这个模块的功能或者这个模块中的蛋白共同参与了该Pathway。P-value的计算为
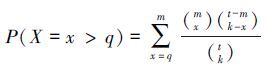 |
(6) |
式中:k代表模块中蛋白的数量,q是模块中被注解的蛋白质数量,m是整个网络中的蛋白质的数量。
1.4 模块的同质性分析一个蛋白质可能被多个GO术语注解,同时一条GO术语也会注解多个蛋白质。一个模块中的蛋白经常会出现多个GO术语描述其功能,因此使用同质性去衡量模块内所有的蛋白质相互作用产生的生物功能的强弱,同质性高则说明该模块内的蛋白质的功能越相近,其计算公式为
 |
(7) |
式中:Gi代表模块中有GO注解的蛋白质的数量,Gij代表模块中共享同一个GO术语的蛋白的数量。
2 融合蛋白质复合体的功能模块检测 2.1 数据的来源及整理STRING 9.1[9] 提供了蛋白质与蛋白质相互作用关系的数据,该数据中包含了一些通过生物实验获得的数据,也包括一些使用计算方法预测出来的数据并使用Score值量化。为了提高PPI网络数据的可靠性,筛选出与人类有关且Score大于700的蛋白相互作用数据,然后将蛋白编码转换为NCBI中名称,最终得到的PPI网络里包括14 380个蛋白质和218 163条蛋白质相互作用。
CORUM[10]存储的是哺乳动物组织器官内经过人工审核过的蛋白质复合体数据,这些数据都是通过个体实验获取的,所以数据噪声少并且准确度高。蛋白质复合体是具有相同功能的蛋白质高度交互的集合,具有较强的生物特性。而蛋白质复合体本身是PPI的一部分,因此将蛋白质复合体数据引入到PPI中,可以弥补其相互作用数据少并且存在噪声的缺陷。本文提取了1 653个与人类相关的蛋白质复合体数据,并且形成了31 550条蛋白质相互作用数据。
2.2 融合蛋白质复合体的PPI网络模块检测将从蛋白质复合体数据中抽取的31 550条蛋白质相互作用数据融入到PPI网络中,从而在一定程度上弥补了PPI数据不足的缺点。由于从蛋白质复合体数据中抽取的这些数据具有很高的精确度,融入这些数据后可以在一定程度上减少PPI中的噪声数据对后续分析的影响。
主要是将抽取到的蛋白质之间的相互作用数据融入到从String9提取的蛋白网络对应的邻接矩阵A中,具体融入方法参照Zhang等[11]提出的方式,将从蛋白复合体中提取出的蛋白质互作数据集合记为C,然后通过融合C和A得到新的邻接矩阵:
 |
(8) |
式中:w是权值,本文中取值为2,融合过程如图 1所示。然后根据新得到的邻接矩阵

|
| 图 1 蛋白互作网络生成过程 Fig. 1 The generation process of protein-protein network |
算法1 蛋白模块检测算法
1) 输入 A, String9.1对应的邻接矩阵;
2) for i= 1:N //每一行代表一个数据点的属性
 |
3) 输出 Gnew:每个蛋白质对应的类标号
算法1将融合了蛋白质复合体的PPI网络划分为K个模块,图 2是分别使用NMF和K-Means社团检测算法检测到的模块238与模块76的拓扑结构图。

|
| 图 2 模块238和模块76的拓扑结构 Fig. 2 The topological structure of module 238 and module 76 |
图 2中节点的名字就是PPI中蛋白质在NCBI中对应的名字,这个名字是唯一的,本文中就是根据这个名字将从Sring9数据中抽取到的PPI同蛋白质复合体数据融合到了一起。可以看出检测到的模块在内部的连接比较紧密。接下来对使用算法1检测到的拓扑模块进行生物学意义上的分析。
2.3 模块的富集分析及同质性分析1) GO术语和Pathway富集结果
对原始的PPI网络和通过融合蛋白质复合体之后的新网络分别进行模块检测,然后对这些模块进行富集分析。为了更好地反应模块的富集结果及同质性,只考虑个数多于2的模块,因为个数为2的模块就只包含一条边,容易对富集结果产生噪声。通过对原始的PPI网络和融合蛋白质复合体的网络分别使用K-Means和NMF对其进行模块划分,并筛选出模块个数大于2的模块,最终检测结果如表 1所示。
| 模块检测算法 | 模块个数 | 最小模块 | 最大模块 |
| K-Means | 266 | 3 | 8 122 |
| IncreK-means | 277 | 3 | 8 157 |
| NMF | 301 | 3 | 307 |
| IncreNMF | 300 | 3 | 328 |
从表 1 可以观察到K-Means算法容易产生比较大的模块,其蛋白质的规模约占整个网络的56%,一般来说这种规模比较大的模块对蛋白质的生物功能分析意义不是很大,而且模块个数在10以下的模块占所有模块的27%左右;而NMF算法检测到的最大模块的规模只占PPI网络的2.28%,而且模块规模小于10的模块占所有模块的比率只有10%,更容易检测到相对规模较中等的模块,更容易获得比较统一的生物功能。
基因本体联合建立了一套适用于不同物种的语义词汇标准,该标准对蛋白质功能等方面进行限定及描述,该标准能够随着研究的深入和时间的发展而不断完善。GO[12]术语就是这个不断增长完善的语义词汇标准的数据库,主要对基因和蛋白质进行注释并且进一步阐明了蛋白质和用于定义它们的GO术语之间的关系。GO术语是生物过程(biological process,BP)、细胞组件(cellular component,CC)和分子功能(molecular function,MF)。每个种类都是一种树形结构,我们总共抽取了40 848条GO术语,其中生物过程有26 958条、细胞组件有3 653条、分子功能包括10 697条。
根据式(6) 对每个模块根据GO术语的3个种类分别进行了富集分析,也就是为每一个蛋白质拓扑模块进行了p-value值的计算,然后选取最小的p-value值对应的GO术语作为该模块的生物功能描述,从而确定该模块中的生物功能。
为了方便比较融合蛋白质复合体数据后检测到的模块与原始PPI网络检测到的模块之间的GO术语富集情况,分别使用GO术语的3个类别对应的所有的GO术语,使用K-Means和NMF两种算法对原始PPI网络和融合了蛋白质复合体的PPI网络划分的模块进行了富集分析,然后对比分析结果。实验表明,融合了蛋白质复合体后划分得到的模块在GO术语上的富集程度要比直接使用原始PPI网络的模块富集程度有显著的提升。
表 2列举了4种方法对应的前20个最小的模块富集结果,分别从生物过程、细胞组件和生物功能3个方面罗列了实验结果,可以看到融合了蛋白质复合体之后的PPI网络得到的模块,在富集程度上比原始模块的p-value值要低,这说明模块的富集程度更好,融合蛋白质复合体的模块更具有显著生物功能上的意义。
| K-Means | IncreK-Means | NMF | IncreNMF | ||||||||
| BP | CC | MF | BP | CC | MF | BP | CC | MF | BP | CC | MF |
| 0.0×100 | 9.4×10-175 | 0.0×100 | 0.0×100 | 2.3×10-176 | 0.0×100 | 0.0×100 | 3.4×10-193 | 0.0×100 | 0.0×100 | 1.9×10-207 | 0.00×100 |
| 5.5×10-104 | 1.79×10-78 | 5.33×10-105 | 4.9×10-104 | 1.20×10-52 | 3.9×10-106 | 1.12×10-48 | 8.49×10-55 | 4.85×10-51 | 2.73×10-49 | 1.00×10-50 | 5.86×10-43 |
| 2.79×10-66 | 2.13×10-48 | 1.45×10-68 | 6.03×10-59 | 1.94×10-48 | 3.87×10-52 | 2.44×10-46 | 3.05×10-52 | 1.25×10-41 | 4.64×10-39 | 2.00×10-44 | 1.28×10-42 |
| 1.53×10-56 | 3.75×10-46 | 5.63×10-43 | 1.24×10-48 | 6.97×10-44 | 1.54×10-48 | 9.89×10-38 | 1.55×10-43 | 3.19×10-38 | 1.93×10-38 | 8.43×10-28 | 3.66×10-32 |
| 3.49×10-50 | 3.80×10-46 | 5.95×10-43 | 2.37×10-41 | 6.99×10-41 | 4.27×10-38 | 3.11×10-36 | 1.02×10-28 | 5.47×10-32 | 2.35×10-38 | 9.42×10-28 | 4.10×10-25 |
| 1.50×10-41 | 9.70×10-31 | 9.20×10-37 | 2.39×10-41 | 1.55×10-29 | 1.45×10-36 | 1.12×10-34 | 3.30×10-28 | 5.46×10-25 | 3.23×10-36 | 2.20×10-27 | 1.61×10-24 |
| 6.73×10-41 | 1.27×10-25 | 1.54×10-31 | 7.20×10-41 | 5.13×10-28 | 1.12×10-33 | 1.39×10-34 | 2.23×10-26 | 1.79×10-24 | 1.25×10-33 | 1.27×10-26 | 1.76×10-24 |
| 3.43×10-39 | 5.71×10-25 | 2.60×10-29 | 8.23×10-41 | 1.12×10-27 | 2.26×10-33 | 1.48×10-31 | 5.26×10-24 | 4.14×10-23 | 3.02×10-33 | 1.35×10-26 | 2.45×10-24 |
| 7.94×10-38 | 1.02×10-24 | 2.94×10-27 | 4.44×10-40 | 4.89×10-26 | 2.77×10-27 | 2.01×10-31 | 1.55×10-23 | 2.99×10-22 | 7.83×10-32 | 1.38×10-26 | 4.95×10-23 |
| 2.17×10-35 | 1.05×10-24 | 5.71×10-27 | 1.71×10-35 | 6.19×10-26 | 1.25×10-26 | 1.38×10-30 | 3.29×10-23 | 1.38×10-19 | 7.97×10-31 | 8.45×10-26 | 1.05×10-22 |
| 3.22×10-35 | 1.67×10-24 | 4.51×10-23 | 2.94×10-35 | 7.81×10-26 | 6.73×10-24 | 2.52×10-29 | 2.78×10-22 | 2.07×10-18 | 1.68×10-29 | 5.48×10-25 | 3.18×10-22 |
| 1.97×10-30 | 2.54×10-24 | 6.86×10-23 | 1.34×10-34 | 2.42×10-24 | 9.83×10-23 | 2.98×10-22 | 2.97×10-22 | 2.10×10-18 | 1.04×10-25 | 7.57×10-25 | 2.21×10-20 |
| 1.76×10-28 | 2.79×10-23 | 2.26×10-21 | 1.85×10-31 | 2.69×10-24 | 3.77×10-22 | 4.38×10-22 | 3.30×10-21 | 4.18×10-18 | 1.34×10-24 | 1.20×10-24 | 2.50×10-20 |
| 1.17×10-27 | 3.16×10-23 | 4.85×10-21 | 4.43×10-31 | 2.05×10-23 | 4.76×10-22 | 4.71×10-22 | 3.80×10-21 | 1.02×10-17 | 1.46×10-24 | 2.04×10-23 | 1.59×10-19 |
| 5.06×10-27 | 6.54×10-22 | 3.03×10-20 | 1.15×10-27 | 2.76×10-23 | 1.84×10-21 | 6.08×10-22 | 6.65×10-21 | 1.20×10-17 | 1.94×10-24 | 2.90×10-23 | 1.17×10-18 |
| 1.80×10-25 | 6.00×10-21 | 2.98×10-18 | 1.83×10-27 | 4.14×10-21 | 2.35×10-21 | 7.70×10-21 | 1.02×10-19 | 4.77×10-17 | 4.55×10-22 | 5.77×10-23 | 1.92×10-18 |
| 2.75×10-27 | 9.72×10-21 | 2.31×10-16 | 2.69×10-27 | 5.63×10-21 | 3.44×10-21 | 1.17×10-20 | 1.12×10-19 | 1.26×10-16 | 9.95×10-22 | 5.57×10-22 | 1.16×10-17 |
| 1.47×10-25 | 1.15×10-18 | 4.27×10-16 | 4.64×10-27 | 1.63×10-19 | 3.87×10-19 | 1.54×10-20 | 1.42×10-19 | 3.92×10-16 | 1.14×10-21 | 7.88×10-22 | 1.76×10-17 |
| 2.46×10-23 | 1.92×10-18 | 6.56×10-16 | 9.92×10-27 | 4.42×10-19 | 1.08×10-18 | 3.36×10-20 | 2.06×10-19 | 6.25×10-16 | 1.99×10-21 | 3.93×10-21 | 5.89×10-17 |
| 1.19×10-22 | 1.94×10-18 | 6.63×10-16 | 8.00×10-25 | 6.78×10-19 | 5.57×10-18 | 3.92×10-20 | 1.02×10-18 | 6.69×10-16 | 2.06×10-21 | 1.38×10-20 | 7.71×10-17 |
同GO的富集分析一样,我们也对模块中蛋白质在Pathway上进行了相应的富集分析,主要是统计一个模块内的蛋白质参与同一条Pathway的程度。Pathway数据主要使用PID[13](pathway interaction database),该数据库由NCI-Nature、BioCarta和Reactome3个数据库整合而成。在本文中只使用分子类型为“蛋白质”和“蛋白质复合体”的数据。最终提取了1 513条Pathway数据,其中223条来自NCI-Nature数据库、254条来自BioCarta数据库、838条来自Reactome数据库。表 3列举了4种方法中对应的前20个最小的模块在Pathway上的富集结果,从中可以看到融合了蛋白质复合体之后的PPI网络的模块,在Pathway上的富集程度比原始的模块的p-value值要低,这说明模块内的蛋白质更多地参与了同一条Pathway,从而可以证明融合了蛋白质复合体的模块更倾向于在同样的Pathway中发挥生物作用,识别Pathway可以帮助人们进一步认识蛋白分子之间相互作用的分子机理。
| K-Means | IncreK-Means | NMF | IncreNMF |
| 8.27×10-41 | 2.58×10-42 | 1.60×10-30 | 1.43×10-38 |
| 8.63×10-41 | 5.23×10-41 | 2.63×10-23 | 4.29×10-27 |
| 7.00×10-33 | 2.48×10-40 | 4.15×10-23 | 1.18×10-26 |
| 2.46×10-30 | 4.16×10-34 | 2.89×10-22 | 7.79×10-23 |
| 1.05×10-22 | 6.62×10-25 | 3.12×10-22 | 8.00×10-23 |
| 3.09×10-19 | 5.38×10-20 | 1.02×10-21 | 1.64×10-22 |
| 5.41×10-18 | 1.06×10-17 | 2.22×10-20 | 6.98×10-18 |
| 5.89×10-18 | 1.95×10-17 | 1.52×10-16 | 5.22×10-17 |
| 1.53×10-17 | 3.18×10-17 | 2.60×10-15 | 1.08×10-16 |
| 1.72×10-16 | 4.47×10-17 | 6.42×10-15 | 1.22×10-15 |
| 2.94×10-15 | 1.59×10-16 | 3.75×10-14 | 8.31×10-15 |
| 1.03×10-14 | 8.01×10-16 | 2.61×10-13 | 2.61×10-14 |
| 1.90×10-14 | 4.45×10-15 | 2.68×10-13 | 6.34×10-14 |
| 9.56×10-14 | 1.82×10-14 | 1.19×10-12 | 1.64×10-13 |
| 2.54×10-13 | 7.16×10-14 | 1.09×10-11 | 6.62×10-13 |
| 1.03×10-12 | 9.74×10-14 | 6.33×10-11 | 2.34×10-12 |
| 2.11×10-12 | 1.33×10-13 | 1.32×10-10 | 1.02×10-11 |
| 2.20×10-12 | 4.10×10-13 | 2.66×10-10 | 2.26×10-11 |
| 5.14×10-12 | 4.10×10-13 | 3.12×10-10 | 2.30×10-11 |
| 1.18×10-11 | 6.00×10-13 | 4.02×10-10 | 2.50×10-11 |
2) 蛋白质拓扑模块同质性
对每个模块使用最小的p-value对应的GO术语或者Pathway作为其富集的对象,从而进一步发现该模块中的蛋白质分子的功能。从统计学意义上讲,p-value < 0.01的GO术语及Pathway都可以作为模块的富集对象。为了更好地衡量模块中的蛋白质在生物功能上发挥相同或相似功能的程度,使用同质性去衡量,其计算方法如式(7) 所示。同质性更好地说明了一个模块内的蛋白在功能上的相似程度,同质性越高说明该模块中的蛋白质在生物功能上更趋于一致性,也就是该模块具有很强的生物功能。
本文对比了融入蛋白质复合体数据之后PPI网络划分得到的模块与原始PPI划分得到的模块之间的同质性的差别。GO术语同质性根据生物过程、细胞组件和分子功能3个方面进行分析。图 3是不同模块划分方法产生模块的分子功能的同质性在不同区间上的对比。

|
| 图 3 模块分子功能同质性 Fig. 3 Molecular function homogeneity of module |
图 3横坐标是同质性区间,纵坐标是该区间内的模块数量占所有模块数量的比率。不论是K-Means还是NMF,融合了蛋白质复合体数据的模块在分子功能的同质性方面要高于原始PPI得到的模块。在K-Means算法中,融合了蛋白质复合体数据的模块中同质性高于0.5的模块占15%,而原始PPI模块同质性高于0.5的模块占11%;在NMF中,融合了蛋白质复合体数据的模块中同质性高于0.5的模块占13%,而原始PPI模块同质性高于0.5的模块占9.6%。
图 4是不同模块划分方法产生模块的细胞组件同质性在不同区间上的对比。

|
| 图 4 模块细胞组件同质性 Fig. 4 Cellular component homogeneity of module |
在K-Means算法中,融合了蛋白质复合体数据的模块中细胞组件同质性高于0.5的模块占54.8%,而原始PPI模块同质性高于0.5的模块占48.9%;在NMF中,融合了蛋白质复合体数据的模块中细胞组件同质性高于0.5的模块占35%,而原始PPI模块同质性高于0.5的模块占31.5%。
图 5是不同模块划分方法产生模块的生物过程同质性在不同区间上的对比。

|
| 图 5 模块生物过程同质性 Fig. 5 Biological process homogeneity of module |
在K-Means算法中,融合了蛋白质复合体数据的模块中生物过程同质性高于0.5的模块占24.1%,而原始PPI模块同质性高于0.5的模块占17.7%;在NMF中,融合了蛋白质复合体数据的模块中生物过程同质性高于0.5的模块占15.7%,而原始PPI模块同质性高于0.5的模块占11.3%。
图 6是不同模块划分方法产生模块的Pathway同质性在不同区间上的对比。
在K-Means算法中,融合了蛋白质复合体数据的模块中Pathway同质性高于0.5的模块占22.3%,而原始PPI模块同质性高于0.5的模块占18.7%;在NMF中,融合了蛋白质复合体数据的模块中Pathway同质性高于0.5的模块占19%,而原始PPI模块同质性高于0.5的模块占12%。
实验结果说明,在GO术语和Pathway2个生物度量方面,不论是从最小富集角度还是从模块同质性角度,都可以发现融合了蛋白质复合体后的PPI得到的模块具有更强的生物功能,因此可以将这些模块作为功能模块,以便用于蛋白网络分子作用机理的研究。

|
| 图 6 模块Pathway同质性 Fig. 6 Pathway homogeneity of module |
本文将蛋白质复合体数据融合到PPI网络中(例如:String 9蛋白质相互作用数据库),然后使用K-Means和NMF 2种经典的算法分别对原始网络和融合后的网络进行社团划分,从而得到多个蛋白质模块;这些模块通过在GO和Pathway2个方面的富集分析和同质性分析,实验结果证明融合蛋白质复合体后得到了生物功能更强的模块;这也在一定程度上说明引入蛋白质复合体数据弥补了PPI网络数据不完整并且噪声多等缺点。新划分的模块在GO和Pathway 2个方面都展现了良好的生物学统计特性,这说明结合多方面的数据,有助于发现功能更强的蛋白质模块。
鉴于目前的研究,下一步工作计划将基因表达数据融入到PPI网络中,然后根据不同的基因在不同组织上的表达情况来辅助PPI网络进行功能模块检测。另一方面,疾病-症状关系数据(OMIM)和疾病-基因关系数据(disease-connect)的获取技术发展比较迅速并且具有较高的可信度, 因此可以将这些数据融入到PPI网络中去发现与疾病或症状相关的功能模块,从而为疾病机理研究和新药研发提供一个新的思路。
| [1] | BARABÁSI A L, GULBAHCE N, LOSCALZO J. Network medicine:a network-based approach to human disease[J]. Nature reviews genetics , 2011, 12 (1) : 56-68 DOI:10.1038/nrg2918 |
| [2] | BADER G D, HOGUE C W V. An automated method for finding molecular complexes in large protein interaction networks[J]. BMC bioinformatics , 2003, 4 : 2 DOI:10.1186/1471-2105-4-2 |
| [3] | ALTAF-UL-AMIN M, SHINBO Y, MIHARA K, et al. Development and implementation of an algorithm for detection of protein complexes in large interaction networks[J]. BMC bioinformatics , 2006, 7 : 207 DOI:10.1186/1471-2105-7-207 |
| [4] | KENLEY E C, CHO Y R. Detecting protein complexes and functional modules from protein interaction networks:A graph entropy approach[J]. Proteomics , 2011, 11 (19) : 3835-3844 DOI:10.1002/pmic.v11.19 |
| [5] | MENCHE J, SHARMA A, KITSAK M, et al. Uncovering disease-disease relationships through the incomplete interactome[J]. Science , 2015, 347 (6224) : 1257601 DOI:10.1126/science.1257601 |
| [6] | NEWMAN M E J. Fast algorithm for detecting community structure in networks[J]. Physical review e , 2004, 69 (6) : 066133 DOI:10.1103/PhysRevE.69.066133 |
| [7] | WAGSTAFF K, CARDIE C, ROGERS S, et al. Constrained k-means clustering with background knowledge[C]//Proceedings of the Eighteenth International Conference on Machine Learning. San Francisco, CA, USA:Morgan Kaufmann Publishers Inc., 2001:577-584. |
| [8] | LEE D D, SEUNG H S. Learning the parts of objects by non-negative matrix factorization[J]. Nature , 1999, 401 (6755) : 788-791 DOI:10.1038/44565 |
| [9] | TURANALP M E, CAN T. Discovering functional interaction patterns in protein-protein interaction networks[J]. BMC bioinformatics , 2008, 9 : 276 DOI:10.1186/1471-2105-9-276 |
| [10] | RUEPP A, WAEGELE B, LECHNER M, et al. CORUM:the comprehensive resource of mammalian protein complexes-2009[J]. Nucleic acids research , 2010, 38 (S1) : D497-D501 |
| [11] | ZHANG Z Y. Community structure detection in complex networks with partial background information[J]. EPL (europhysics letters) , 2013, 101 (4) : 48005 DOI:10.1209/0295-5075/101/48005 |
| [12] | ASHBURNER M, BALL C A, BLAKE J A, et al. Gene Ontology:tool for the unification of biology[J]. Nature genetics , 2000, 25 (1) : 25-29 DOI:10.1038/75556 |
| [13] | SCHAEFER C F, ANTHONY K, KRUPA S, et al. PID:the pathway interaction database[J]. Nucleic acids research , 2009, 37 (S 1) : D674-D679 |