
 2017, Vol. 37
2017, Vol. 37文章信息
- 梁坚坤, 李涛洪, 曹明, 席雪冬, 吴志刚, 杜官本
- LIANG Jiankun, LI Taohong, CAO Ming, XI Xuedong, WU Zhigang, DU Guanben
- 碱催化三聚氰胺甲醛体系中的竞争关系
- Study on the competitive relationships of the base-catalytic melamine formaldehyde system
- 森林与环境学报,2017, 37(2): 251-256.
- Journal of Forest and Environment,2017, 37(2): 251-256.
- http://dx.doi.org/10.13324/j.cnki.jfcf.2017.02.021
-
文章历史
- 收稿日期: 2016-06-03
- 修回日期: 2016-09-09




2. 西南林业大学云南省木材胶粘剂与胶合制品重点实验室, 云南 昆明 650224
2. Key Laboratory of Wood Adhesives and Adhesive Products, Southwest Forestry University, Kunming, Yunnan 650224, China
三聚氰胺 (melamine,M) 是一种很重要的有机化工原料,其可以与甲醛 (formaldehyde,F) 缩聚生产三聚氰胺甲醛 (melamine formaldehyde,MF) 树脂,属于氨基树脂的一种类型[1]。与尿素 (urea,U) 的结构类似,两者活性官能团都是氨基 (—NH2),但M存在三嗪环大π键,并且M上含有对称的3个氨基,正是由于M结构的特殊性,MF相对于脲醛 (urea formaldehyde,UF) 树脂具有更好的耐水性和耐候性,胶接强度高,热稳定好, 而缺陷在于储存稳定性较差,脆性较高,成本较UF要高很多。以往对UF在不同条件下的亚甲基桥键和醚键的竞争反应研究[2]较多,但对于MF在反应过程中的竞争历程少有研究。
目前关于MF树脂的基础研究主要是早期的一些反应动力学研究[3-4]和结构表征[5-6]以及后期固化过程[6]和树脂储存稳定性的研究[7],对于反应过程中的竞争反应少有报道。可能是MF树脂的合成工艺简单,反应条件对树脂结构性能的影响比较简单所致。但实际上MF树脂合成过程中竞争反应的演变机理还不是很清楚,结构与反应条件之间的关系还不是很清晰。
电喷雾电离质谱仪 (electrospray ionization mass spectrometer,ESI-MS) 和核磁共振碳谱 (13C-nuclear magnetic resonance,13C-NMR)[2, 6, 8]是目前研究结构过程比较行之有效的主要手段。借用这2种技术手段对MF反应体系中的竞争关系进行探讨,对其中的反应机理进行合理解释。
1 试验样品制备与表征 1.1 试验原料M (分析纯/99%)、F (分析纯/37%~40%),国药集团出品,氢氧化钠 (分析纯/99%),市售。
1.2 样品制备pH值为8.0~9.0,F/M的摩尔比为2,分别在50、60、70、80 ℃水浴温度下制备样品,反应时间1 h,每个温度样品依次编号为LA1、LA2、LA3、LA4,冷却备ESI-MS测定用。在pH值为8.0~9.0,90 ℃水浴温度下,分别制备F/M的摩尔比为1、2和3,反应时间1 h,分别取样编号为MFB1、MFB2、MFB3,冷却备13C-NMR测定用。
1.3 仪器表征电喷雾电离质谱仪 (ESI-MS):Waters公司生产的XEVO-TQS#WAA164型质谱仪。离子源为ESI+;质量分析器的低端分辨率/高端分辨率为2.9/14.3;离子能量为0.3 eV;进样速度为5 μg·s-1。图谱处理软件为Waters质谱仪自配的质谱数据软件和电脑画图软件。核磁共振碳谱 (13C-NMR) 仪:Bruker-AVANCE 600超导核磁共振仪。共振频率为150 MHz,将300 μL液体样品与100 μL氘代二甲亚砜溶剂 (DMSO-d6) 混合均匀作为待测样品。测定条件:弛豫时间6 s;反转门控去耦法;zgig脉冲程序;扫描累加次数为800~1 200次。谱图中亚甲基碳均来自于甲醛或甲二醇,每种亚甲基碳的积分面积与总亚甲基碳面积的商值即为各亚甲基碳含量占总亚甲基碳的比例。图谱处理为超导核磁共振仪自配软件和电脑画图软件。
2 分析与讨论在MF碱性体系中涉及的反应类型与UF类似,首先发生羟甲基化,三聚氰胺羟甲基化机理为
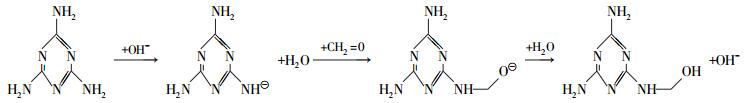
|
这一机理是OKANO et al[9]在20世纪50年代提出的理论,SATO et al[3]对这机理路线提出了疑问,认为M显弱碱性 (pH=8),很难与碱反应生成M负离子 (较强碱性),弱碱制备强碱违背基本化学常识。但计算表明M-负离子与F的反应能垒为6.4 kJ·mol-1,比扩散控制反应如酸碱中和反应能垒 (13~25 kJ·mol-1) 小很多。根据阿伦尼乌斯反应速率方程,由于M-与F的反应能垒很低,反应速率常数比扩散控制反应速率大,平衡反应可以正方向移动。实际上M显弱碱性的原因现在还未定论,大部分认为是处于间位的三游离氨基质子化而显碱性,这与氨基电离出质子形成M负离子过程相反;而从M的结构出发,很显然三嗪环上的N上存在未配位电子对以及环外氨基电子存在离域现象使得环N电子云密度增大,更容易质子化,同时环外游离氨基上氮原子未配位电子与环存在p-π共轭,使得氨基N—H极性增强,显弱酸性,离去H+生成氨基负离子电荷可以较大程度分散到大π环上,负离子有相对稳定性。综上分析OKANO的机理存在合理性。在此基础上对相关反应过程进行了仪器分析。
2.1 电喷雾电离质谱仪分析图 1显示的是LA1~LA4样品的ESI-MS图谱,图 2表示的是其中缩聚产物部分比较图。根据相关文献研究,M在纯水中的溶解度很低,LA1~LA4反应体系中开始有部分不溶M,而固-液两相之间反应速率很慢,未溶解的M几乎不参与反应,溶解的M与F发生亲核加成反应,使M的溶解平衡向正方向移动。

|
图 1 样品LA1、LA2、LA3和LA4的ESI-MS图 Fig. 1 The ESI-MS of LA1, LA2, LA3 and LA4 samples |

|
图 2 样品LA1、LA2、LA3和LA4缩聚产物的ESI-MS图 Fig. 2 The ESI-MS of the condensation products of L-A1, L-A2, L-A3 and L-A4 samples |
根据质谱的测试原理[11],M的结构上含有3个间位的—NH2基团,易与H+结合离子化,图 1中质荷比127是[M+H]+,139是[M=CH2+H]+,151是[CH2=M=CH2+H]+。但在MF碱性合成反应体系中形成M=CH2结构难度很大,且在下节碳谱中也没有出现N=CH2(化学位移在140×10-6~150 ×10-6之间) 的双键结构也证明了这一点。而在ESI-MS谱图中很明显的出现这种结构,这与ESI-MS的检测原理有关。质谱仪在出峰前需要先将体系结构离子化,而一般碱性环境使用正离子检测模式,ESI离子源提供参与离子化较普遍的是H+和Na+。实际上质荷比139峰是由一羟甲基三聚氰胺 (MF1) 在质谱仪检测环境中产生,产生机理为

|
这结构以H+归属为质荷比139,以Na+归属则为质荷比161,在图 1各个样品图中均有出现,这也说明羟甲基三聚氰胺在酸性条件下不稳定。同理,质荷比151对应的结构是CH2=M=CH2(H+)。根据文献[12],主要结构归属见表 1。
| 测量值 Measured value |
化学结构Chemical structure | |
| [M+H]+ | [M+Na]+ | |
| 127 | _ | M |
| 139 | 161 | M=CH2 |
| _ | 191 | M (—CH2OH)=CH2 |
| 151 | _ | CH2=M=CH2 |
| _ | 179 | MCH2OH |
| _ | 209 | M (—CH2OH)2 |
| _ | 239 | M (—CH2OH)3 |
| _ | 269 | M (—CH2OH)4 |
| - | 347 | M (—CH2OH) CH2M (—CH2OH) |
| M (—CH2OH)3CH2OCH2M | ||
| - | 377 | M (—CH2OH) CH2M (—CH2OH)2 |
| M (—CH2OH) CH2OCH2M (—CH2OH) | ||
| M (—CH2OH)2CH2OCH2M | ||
| MCH2M (—CH2OH)3 | ||
| - | 407 | M (—CH2OH) CH2M (—CH2OH)3 |
| M (—CH2OH)2CH2OCH2M (—CH2OH) | ||
| M (—CH2OH)3CH2OCH2M | ||
| M (—CH2OH)2CH2OCH2M (—CH2OH) | ||
| MCH2M (—CH2OH)4 | ||
| M (—CH2OH) CH2M (—CH2OH)3 | ||
| M (—CH2OH)2CH2M (—CH2OH)2 | ||
| - | 455 | MCH2MCH2M (—CH2OH) |
| - | 485 | MCH2OCH2MCH2M (—CH2OH) |
| MCH2OCH2MCH2OCH2M | ||
| MCH2M (—CH2OH) CH2M (—CH2OH) | ||
| M (—CH2OH) CH2MCH2M (—CH2OH) | ||
LA1~LA4样品谱图中均出现质荷比151、179、209、239、269,表明M羟甲基化是普遍存在的,形成了一、二、三和四羟甲基三聚氰胺,四羟甲基三聚氰胺 (MF4) 以上的含量很少,几乎没有,可能原因是F/M的摩尔比低,基本不形成MF4以上化合物;MF4结构中存在偕二羟甲基,反应位阻增大,难以形成。从相对含量分析,发现图 1中主要是羟甲基化结构峰,缩聚产物峰明显很少。
从图 2可以看出,随着温度的升高,缩聚产物的含量有所升高,但趋势规律不是很明显,原因在于温度升高使正逆反应速率都提高;反应时间短,反应物浓度低,温度变化的影响并未体现出来;缩聚反应是放热反应,从平衡的角度分析,温度升高逆反应速率会有所增大,不利于缩聚反应。而在相同条件下UF体系发生了较大程度的两分子羟甲基脲缩合,而LA1~LA4样品谱图大部分是羟甲基三聚氰胺,质荷比347、377、407对应的两分子羟甲基三聚氰胺缩合物相对较少。根据理论计算,碱性条件下羟甲基三聚氰胺之间的缩合反应的活化能为112.8 kJ·mol-1,比羟甲基脲之间的缩合活化能132.8~148.8 kJ·mol-1低,低活化能并没有导致缩合产物的增加。究其原因,一个是因为M在纯水中的溶解度太低,导致生成的羟甲基三聚氰胺浓度不够,比相同条件下的羟甲基脲要低,直接使缩合产物浓度偏低,这也是在合成MF树脂时要在高温条件下的重要原因;以往研究都讨论到M的反应活性大,树脂成胶快,实际上M缩聚反应活性不比U快很多,高温条件下合成的MF树脂中羟甲基含量依然很高[13-14](能达到60%~70%),缩聚产物不是很多,另一个原因是M本身分子量比较大,是U的2倍多,缩聚产物分子量较同条件下的UF大,同时,成胶快是一种宏观表象,可能是存在低聚物凝胶聚合和存在氢键静电等超分子现象[15, 13],一定程度上阻碍了缩聚反应。在MF反应体系中,参照表 3,质荷比347、377、407对应的两分子羟甲基三聚氰胺缩合物,OH-离子作为催化剂参与缩聚反应,其机理为
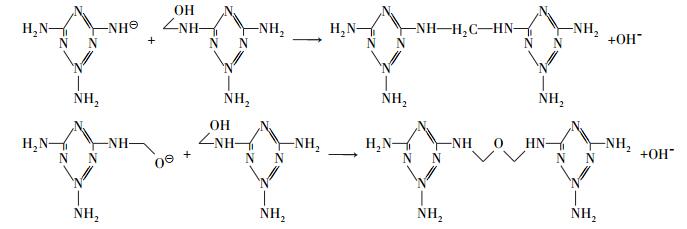
|
以上为SN2催化机理,表明OH-离子首先催化M或MF形成M氨基负离子或羟甲基三聚氰胺氧负离子,再与羟甲基三聚氰胺的羟甲基发生亲核反应,生成亚甲基桥键 (—CH2—) 或醚键 (—CH2OCH2—) 缩聚物。由于负离子浓度很低,氨基负离子或羟甲基负离子与另一羟甲基三聚氰胺之间的反应活性不高。质荷比455、485对应的是三分子羟甲基M缩合反应产物,根据计算, 质荷比455结构中只可能以桥键结合。而对于三聚氰胺缩聚产物中桥键和醚键的竞争关系,在质谱当中体现的不是很明显,表 1中显示出质谱峰对应的结构存在较多的桥键和醚键异构和位置异构,无法准确描述醚键和桥键的竞争关系,对于它们的竞争关系,需要借助核磁共振碳谱进行研究分析。
2.2 核磁共振碳谱分析碱性条件下,MF体系中的反应与UF类似,存在羟甲基化和缩聚两种反应,缩聚产物中以亚甲基桥键和亚甲基醚键2种形式连接,两者是一对竞争反应,由于ESI-MS中普遍存在同分异构体现象,无法准确描述这种竞争关系。而核磁共振定量碳谱中不同的化学位移代表着特定的基团,积分相对大小可以说明竞争关系的强弱。
在合成氨基树脂过程中F/M摩尔比是影响产物分布的一个重要因素,为了说明MF体系中各结构的竞争关系,讨论不同摩尔比条件下产物结构分布情况,使用13CNMR技术手段进行分析。根据参考文献[7, 13],MF体系中化学位移在 (68~80)×10-6为链型亚甲基醚键结构,在 (46~48)×10-6为亚甲基桥键,在 (64~65)×10-6为羟甲基三聚氰胺基团,在 (83~92)×10-6对应于甲二醇及其缩聚物,将各样品峰的归属及定量结果列于表 2。
| 化学结构 Chemical structure |
化学位移 (×10-6) Chemical shift |
含量百分比Percentage composition/% | ||
| MFB1(F/M=1) | MFB2(F/M=2) | MFB3(F/M=3) | ||
| —NHCH2OCH2NH—(Ⅰ) | 68~69 | 29.4 | 40.4 | 36.90 |
| —NHCH2OCH2N=(Ⅱ) | 74~76 | - | - | 0.38 |
| =NCH2OCH2N=(Ⅲ) | 79~80 | - | - | - |
| —NHCH2OH | 64~65 | 61.4 | 53.6 | 56.00 |
| —NHCH2OCH3 | 72~73 | 2.1 | 4.5 | 3.15 |
| —NHCH2NH—(Ⅰ) | 46~48 | 6.0 | 0.6 | - |
| —NHCH2N (Ⅱ) | 53~54 | - | - | - |
| NCH2N (Ⅲ) | 60~61 | - | - | - |
| HOCH2OH | 83~84 | 0.4 | 0.3 | 0.77 |
| HOCH2OCH2OH | 87~88 | 0.3 | 0.3 | 1.46 |
| HOCH2OCH2OCH2OH/HOCH2OCH3 | 90~92 | 0.3 | 0.3 | 1.34 |
在表 2中,F/M摩尔比为1时,亚甲基醚键含量为29.4%,而桥键也达到6%,醚键含量是桥键的4.9倍,存在明显竞争关系。此时体系中主要生成的是单羟甲基三聚氰胺,体系当中含有较多的游离氨基,增大了游离氨基与羟甲基三聚氰胺之间碰撞的概率,有利于生成桥键。当F/M摩尔比的升高为2时,游离氨基含量会大幅度下降,缩聚反应主要发生在羟甲基之间,主要生成亚甲基醚键。此时形成桥键,需要克服临近—CH2OH对OH-离子的位阻效应,难度增大,表 2中显示醚键与桥键的比例从之前的4.9升到67.33,印证了这一点。当摩尔比达到3时,已经观察不到桥键信号,表明摩尔比对缩聚产物的分布影响很大。总的表明,在低F/M摩尔比下,桥键和醚键的形成反应存在明显的竞争关系,随着F/M摩尔比的升高,亚甲基醚键的形成更具优势。
相比于UF碱性体系,研究[12]表明F/U为1时,同样条件下亚甲基桥键可占到醚键80%左右,而以上实验显示MF体系中却不到20%。从统计学分析,M结构当中含有3个游离氨基,同样摩尔比下,MF体系中游离氨基含量更大,生成桥键的概率应该不低于UF体系,而实际上MF体系生成的桥键更少。很显然的一个原因是生成醚键所需要的反应能垒更低,这是桥-醚键竞争关系的重要解释,无法合理解释在同样条件下MF体系中桥键与醚键的相对比例小于UF体系。实际上树脂合成反应当中反应动力学控制速率,热力学决定产物的分布,产物的能量越低越有利于形成。从能量上分析,根据理论计算UF体系中桥键是比醚键能量更低更稳定的结构,所以在反应的最终状态时桥键要远远大于醚键,但由于M存在大π环特殊结构,经过计算MF体系中亚甲基醚键结构比桥键能量更低,这与UF体系正好相反。进一步研究发现,亚甲基醚键结合的MF缩聚物容易使得两个大π环在空间上平行折叠,产生了超分子化学当中的π-π堆积效应,使得体系更为稳定。这对于MF体系当中生成醚键更具优势提供了一个合理解释。
在表 2中,羟甲基含量达到53%以上,而合成的MF初级树脂[12]中更是可能高达70%~85%,甲醛参与的缩聚程度没有UF体系高,说明缩聚反应程度有限,以往认为M反应活性大,更多的在于羟甲基化活性,在缩聚反应中羟甲基三聚氰胺的反应活性并不是很大。碱性条件下体现出的成胶快并不等价于反应快,成胶是一种胶体宏观现象,与凝胶聚合和氢键等等有关[13]。同时M本身具有支链结构,在树脂化过程中易于支链化而导致成胶快。但对于羟甲基三聚氰胺缩聚反应速率慢的原因还需要进一步深入地研究。
3 结论MF碱性体系中,OH-离子催化形成三聚氰胺氨基负离子和羟甲基三聚氰胺氧负离子参与羟甲基化和缩聚反应,MF体系中主要发生羟甲基化,缩聚产物较少。碱性条件下,在低F/M摩尔比下,桥键和醚键的形成反应存在明显的竞争关系,随着F/M摩尔比的升高,亚甲基醚键的形成更具优势。MF体系中醚键结合的缩聚产物易于在空间上产生平行折叠,形成π-π堆积超分子效应,能量更低,有利于形成。M反应快主要在于羟甲基化,MF碱性条件下成胶快是一种宏观表象,树脂结构中羟甲基含量占主要部分,缩聚产物含量不大。成胶的原因在于易于形成氢键络合、凝胶聚合,以及本身较差的水溶性。
| [1] | 熊冕, 马迎春, 王姣姣, 等. 三聚氰胺甲醛树脂研究与应用进展[J]. 广东化工, 2013, 40(22): 68, 74. DOI:10.3969/j.issn.1007-1865.2013.22.035 |
| [2] | 韩书广, 吴羽飞. 脲醛树脂化学结构及反应的13C-NMR研究[J]. 南京林业大学学报 (自然科学版), 2006, 30(5): 15–20. |
| [3] | SATO K, KONAKAHARA T, KAWASHIMA M. Studies on formaldehyde resins, general base catalysis in hydroxymethylation of melamine with formaldehyde[J]. Macromolecular Chemistry and Physics, 1982, 183(4): 875–881. |
| [4] | BERGE A, MEJDELL T. Melamine formaldehyde compounds: the active species in acid catalyzed reactions[J]. Polymer, 2006, 47(9): 3249–3256. |
| [5] | MERCERL A T, PIZZI A. A 13C-NMR analysis method for MF and MUF resin strength and formaldehyde emission from wood particleboard: Ⅱ. MF resins[J]. Journal of Applied Polymer Science, 1996, 61: 1697–1702. |
| [6] | 王辉, 杜官本, 王洪艳. 不同合成工艺路线下MUF共缩聚树脂结构分布[J]. 林业科技开发, 2015, 29(3): 97–100. |
| [7] | MERLINE D J, VUKUSIC S, ABDALA A A. Melamine formaldehyde: curing studies and reaction mechanism[J]. Polymer, 2013, 45(4): 413–419. |
| [8] | 席雪冬, 吴志刚, 王辉, 等. 高浓度甲醛制备脲醛树脂及其性能分析[J]. 森林与环境学报, 2015, 35(3): 210–213. |
| [9] | OKANO M, OGATA Y. Kinetics of the condensation of melamine with formaldehyde[J]. Journal of the American Chemical Society, 1952, 74(22): 5728–5 731. |
| [10] | 许迁, 温绍国, 王继虎, 等. 三聚氰胺饱和溶解度与温度和pH值关系研究[J]. 化学世界, 2010, 51(8): 473–475, 455, 458, 490. |
| [11] | 陈耀祖, 涂亚平. 有机质谱原理及应用[M]. 北京: 科学出版社, 2004: 55-57. |
| [12] | 李涛洪. 木材胶粘剂用氨基树脂合成反应机理研究[D]. 南京: 南京林业大学, 2015. |
| [13] | 连海兰, 赵丽艳, 洪枢, 等. 三聚氰胺甲醛树脂的聚合机理及其在液体耐磨技术中的应用[J]. 东北林业大学学报, 2014, 42(8): 115–119, 130. |
| [14] | 邹怡佳, 陈玉和, 吴再兴. 改性三聚氰胺树脂的研究进展[J]. 林产化学与工业, 2013, 33(5): 127–130. |
| [15] | 吴书泓, 罗朝晖, 孙振鸢. 利用C-13 NMR探讨影响MF树脂贮存稳定性的因素[J]. 木材工业, 1998, 12(4): 8–11, 14. |