
文章信息
- 许俊杰, 闫文亮, 赵团结, 姜海燕
- XU Junjie, YAN Wenliang, ZHAO Tuanjie, JIANG Haiyan
- CROPGRO大豆花期模拟模型品种参数的遗传学解析
- Genetic analysis of cultivar parameters of CROPGRO soybean flowering simulation model
- 南京农业大学学报, 2021, 44(4): 778-788
- Journal of Nanjing Agricultural University, 2021, 44(4): 778-788.
- http://dx.doi.org/10.7685/jnau.202007011
-
文章历史
- 收稿日期: 2020-07-06




2. 南京农业大学国家大豆改良中心, 江苏 南京 210095
2. National Center for Soybean Improvement, Nanjing Agricultural University, Nanjing 210095, China
基于过程的作物生长模拟模型(简称作物生长模拟模型)是以作物生长发育的生理规律为基础, 综合作物遗传特性、环境因素和田间管理措施三者相互作用关系的基础上, 利用数学方法定量模拟光合生产、阶段发育、器官建成和产量形成等生理过程的机制模型[1]。作物生长模拟模型通过量化作物每一生理过程中对环境(例如温度、光照等)变量的非线性响应, 将复杂表型性状分解为受单一或复合环境变量和基因型互作影响的品种参数(cultivar parameter, CP), 并以此反映环境对作物品种造成的影响[2]。作物生长模拟模型着重于量化环境效应对目标表型性状造成的影响(过程层面), 通过品种参数体现基因型与环境互作[3]。因此, 生长模拟模型的品种参数应当包含影响对应表型性状的遗传学基础特性。例如, 品种参数可以与数量性状基因座[4]或与候选基因的等位基因信息直接相关[5-6]。单个品种参数应处于简单且独立的遗传控制之下, 并且一组不同的品种参数可以描述某一特定基因型材料的基因特性[7]。但是, 目前作物模型品种参数是根据观测到的表型数据变异获得, 而不是根据群体遗传变异而获得, 造成品种参数与遗传学基础特性之间的联系不够密切[8], 限制了作物生长模型对基因型与环境互作的表达能力。
自1996年, White等[5]将作物模型品种参数定义为“遗传系数”以来, 越来越多的研究者开展了模型品种参数的遗传学可解释性研究[9-12]。Guitton等[10]基于CERES模型将高粱开花时间分解为光周期敏感性、临界光周期等与光反应有关的模型品种参数, 发现在3号染色体的74 cM的位置上, 存在着与所有模型品种参数显著相关的候选基因区间, 可解释高达40%的品种参数变异; Dingkuhn等[11]利用能够模拟水稻温光互作效应的RIDEV V2模型模拟花期, 通过低生长温度、光周期敏感性等6个品种参数进行全基因组关联分析后发现, 品种参数低生长温度定位到的QTL与低温下诱导成花的基因HD3a同源; Kadam等[12]利用GECROS模型将水稻产量性状分解为光周期敏感性(δ)和株高峰值(Hmax)等8个作物模型品种参数, 结果表明, 显著关联的SNP标记可解释42.2%~77.0%的品种参数变异。表明上述模型的品种参数具有遗传效应。利用SNP标记或QTL对作物模型品种参数进行优化, 通过将作物生长模拟模型与基因组选择预测模型连接起来, 构建基因型-环境互作模型。其前提是: 所研究的作物生长模拟模型品种参数具有遗传效应, 并可以通过筛选的显著相关SNP标记加以表达[4]。
大豆[Glycine max(L.)Merr.]为光周期反应的模式作物, 外界的光反应显著影响着大豆花期等重要性状。CROPGRO大豆花期模型可以模拟温光条件变化对大豆花期影响。目前国内外未见对CROPGRO大豆花期模型品种参数遗传特性进行解析的研究报道。本研究以江淮地区183个优质高产大豆品种在多生态点种植的花期数据为材料, 利用多种全基因组关联分析手段对CROPGRO大豆花期模型品种参数进行遗传学解析, 验证其是否具有遗传效应, 为构建基因型-环境互作的大豆花期模拟模型奠定理论基础。
1 材料与方法 1.1 大豆试验材料及田间表型评估以长江江淮地区183个优质高产大豆品种为研究对象, 这些种质资源均来自南京农业大学国家大豆改良中心。试验于2011—2018年, 在江苏南京、盐城、溧水及安徽当涂和河南新乡10个生态点种植, 采取行播种植, 行长2 m, 行宽0.5 m, 共设3个试验重复。当地气象数据来源于中国国家气象局(http://www.nmic.cn/)。
根据Fehr的物候期分类法[13], 供试材料的花期为从播种至一行中50%的大豆植株开花的时间(d), 用SPSS 25.0和Excel 2016软件对表型数据进行基本统计分析, 利用R语言的lmr4[14]包对表型进行最佳线性无偏估计(BLUP)值的计算。
| 环境缩写 Environmental abbreviation |
经纬度 Latitude and longitude |
播种日期 Sowing date |
平均最高温度/℃ Average maximum temperature |
平均最低温度/℃ Average minimum temperature |
平均日照时数/h Average day length from |
| NJ11 NJ13 NJ14 NJ18 |
32°3′32″N, 118°37′40″E | 2011-06-24 2013-06-19 2014-06-21 2018-07-03 |
31.7 34.3 29.9 33.6 |
24.9 26.3 23.3 25.7 |
13.76 13.74 13.69 13.53 |
| YC17 YC18 |
33°22′39″N, 120°8′24″E | 2017-06-30 2018-06-22 |
33.4 32.2 |
26.5 25.1 |
13.76 13.59 |
| LS11 | 31°39′11″N, 119°1′43″E | 2011-07-20 | 31.4 | 24.1 | 13.15 |
| DT11 DT18 |
31°34′15″N, 118°29′52″E | 2011-06-24 2018-07-05 |
31.9 33.5 |
25.1 25.6 |
13.66 13.44 |
| XX18 | 35°18′09″N, 113°53′02″E | 2018-06-16 | 33.6 | 25.4 | 13.95 |
| 注: 1)环境缩写的命名规范是: 地点缩写+年份。DT: 当涂; NJ: 南京; YC: 盐城; LS: 溧水; XX: 新乡。下同。 2)平均最高温度、最低温度和日照时数为每个种植环境下从播种至最后一个大豆品种开花的时间区段内的对应气象数据的平均值。 Note: 1)The naming convention for environmental abbreviations is: place abbreviation+year. DT: Dangtu; NJ: Nanjing; YC: Yancheng; LS: Lishui; XX: Xinxiang. The same as follows. 2)The average maximum temperature, minimum temperature and sunshine duration are the average values of corresponding meteorological data in the time interval from sowing to flowering of the last soybean variety in each planting environment. | |||||
供试大豆通过简化基因组测序技术(restriction-site associated DNA sequencing, RAD-seq)共获得87 307个高通量SNP(single nucleotide polymorphism)数据。参照测序数据的质量控制和调用变异(calling variations)方法[15], 本试验保留最小等位基因频率(minor allele frequency, MAF)大于0.05的变异位点, 最终获得60 712个高质量且位置唯一的SNP标记用于后续分析。
1.3 基于BLUP的花期表型数据集构建及花期模拟模型品种参数校正本研究基于10个环境的实测花期数据, 计算花期最佳线性无偏预测值(best linear unbiased prediction, BLUP)。为最大程度减少不同环境差异对同一品种表型造成的影响, 构建了实测花期和BLUP花期2类数据集, 分别命名为E-FT和B-FT, 利用它们完成后续的CROPGRO大豆花期模拟模型品种参数校正试验, 并将所得品种参数命名为E-CP和B-CP。
1.4 CROPGRO大豆花期模拟模型算法原理CROPGRO大豆花期模拟模型(以下简称CROPGRO-Soybean-Flower模型), 为DSSAT软件[16]中的1个豆科模拟模型, 由BEANGRO模型[17]发展而来, 以大豆发育生理生态为基础, 采用生理发育时间作为定量发育进程的尺度, 以天为步长模拟大豆生长发育。大豆生长累计进度可为定义为:

|
(1) |
式中: FT为每日实际生长光热的累计值; D为实际开花所需要的时间(d); fT(m)为第m天的温度效应; fDL(m)为第m天的光照效应。
FT包括了播种到出苗时长(PLEM)、出苗到开花时长(EMFL)2个物候阶段, 其中PLEM对于大多数品种都相近, 模型将其设为3.6 d。当FT满足公式(2)时, 大豆进入花期。
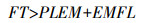
|
(2) |
每日的温度效应和光照效应又可被定义为:

|
(3) |
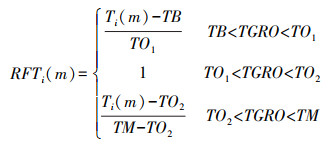
|
(4) |

|
(5) |
式中: RFTi(m)为第m天第i小时的热效应; Ti(m)为第m天第i小时温度; TB为大豆生长率为0时的最低温度; TM为大豆生长率为0时的最高温度; TO1、TO2依次为最适温度下限和上限。在本文中使用模型默认值[18], TB=7 ℃、TO1=28 ℃、TO2=35 ℃、TM=45 ℃。DL(m)为第m天的光照时间; CSDL为临界光周期; PPSEN为光周期敏感性。
由以上公式可以看出, 除了模型设定和田间实测的数据外, CSDL、PPSEN和EMFL为模型待调遗传参数, 取值范围分别为11.78~14.60 h、0.129~0.385 d和15.5~28.9 d。
CROPGRO大豆花期模拟模型的驱动数据输入可分为3种类型: 气象数据、田间管理数据、作物品种参数。气象数据包括日最高气温(℃)、日最低气温(℃)、日照时数(h)、纬度(°)。田间管理数据包括行距(cm)、种植深度(cm)等数据。
1.5 基于GLUE校正大豆花期模拟模型品种参数本研究选用DSSAT-GLUE程序包[19](https://www.dssat.net/)对10个环境下CROPGRO大豆花期模拟模型品种参数进行校准。总采样次数设为20 000, 在品种参数范围内均匀随机采样, 作为品种参数校正的初始值。运行使用的计算机环境是Intel(R)Core(TM)i5-7200U CPU@ 2.50 GHz, 内存8 GB, Windows10 64位操作系统。
1.6 评价指标为了评价品种参数集的质量, 本研究用1∶1图判断模型模拟值与实测值之间的吻合程度。利用模拟结果与实测结果的平均值、标准差(SD)、均方根误差(RMSE)来度量模型的模拟误差。
总体模拟效果由全部品种的均方根误差的均值(ARMSE)来反映, 单位为d:

|
(6) |
式中: N为供试大豆品种数; j表示第j个品种。
1.7 遗传多样性、群体结构和主成分分析利用STRUCTURE v2.3.4软件对关联群体的群体结构进行分析, 即计算群体内所有品种所属的亚群。将群体内存在的亚群数目K值设定为1~11, 迭代次数设置为10 000, 马尔科夫链蒙特卡罗(Markov chain Monte Carlo, MCMC)迭代次数设置为100 000, 针对每个K值, 重复运行7次, 最终通过Evanno等[20]的计算方法计算最优亚群数(ΔK), 并获得Q矩阵。利用TASSEL v5.0软件对供试大豆的遗传多样性进行分析, 绘制邻接树, 以此作为全基因组关联分析的协变量。用GCTA v1.92软件进行种群结构的主成分分析。
1.8 全基因组关联分析为了研究CROPGRO大豆花期模拟模型品种参数的遗传可解释性, 利用STRUCTURE v2.3.4软件计算得到的群体结构Q矩阵和TASSLE v5.0软件计算得到的亲缘关系K矩阵, 采用TASSLE v5.0软件的MLM方法[21]以及mrMLM软件包的mrMLM[22]、FASTmrMLM[23]、FASTmrEMMA[24]、pLARmEB[25]、pKWMEB[26]和ISIS EM-BLASSO[23]模型共7个GWAS分析方法, 基于60 712个高质量SNP标记(MAF>0.05), 分别对花期BLUP值(B-FT)和基于BLUP值分解的3个模型品种参数性状(B-CP)进行关联分析。利用R语言的QQman包对MLM的输出结果进行曼哈顿图和QQ图的绘制[27], 除MLM方法设置显著关联SNP的建议阈值为-lg p≥3.5, 显著阈值设为-lg p-lg(1/60 712)=4.78外, 其余6种方法均选取建议的LOD≥3.0作为SNP显著关联的标准。
1.9 候选基因的功能注释本研究将至少被2种GWAS分析方法检测到的QTN视作稳定关联标记, 用于后续分析。将显著关联QTN上、下游各500 kb[28]设定为候选基因筛选区间, 利用SoyBase(http://www.soybase.org)和TAIR(https://www.arabidopsis.org/)生物信息学网站和前人研究进展对候选基因的功能进行预测。
2 结果与分析 2.1 实测花期数据统计分析在2011—2018年10个种植环境下, 对183种江淮大豆种质花期性状进行统计。表 2结果显示, 10个环境的大豆花期平均值为38.0~48.3 d, 对应的变异系数为7.38%~16.49%, 花期最小值(31 d)和最大值(67 d)分别出现在LS11、YC18和XX18三个环境中, 花期BLUP值的平均值为43.5 d, 变异系数为7.78%。从统计分析来看, 大豆花期性状在不同种植环境下的表型存在着一定差异, 表明开花时间易受环境影响。
| 环境 Environment |
花期/dFlowering period | 变异系数% Coefficient of variation |
偏度 Skewness |
峰度 Kurtosis |
|||
| 均值±标准差 Mean±SD |
最小值 Minimum |
50%分位数 50% quantile |
最大值 Maximum |
||||
| DT11 | 42.4±4.8 | 33 | 41 | 57 | 11.25 | 0.33 | -0.09 |
| NJ11 | 41.0±3.6 | 34 | 40 | 51 | 8.80 | 0.53 | 0.01 |
| LS11 | 38.0±2.8 | 31 | 38 | 50 | 7.38 | 0.66 | 2.57 |
| NJ13 | 45.9±5.3 | 34 | 46 | 61 | 11.50 | 0.24 | -0.28 |
| NJ14 | 46.8±5.0 | 32 | 46 | 62 | 10.79 | 0.15 | 0.85 |
| YC17 | 41.2±4.8 | 32 | 41 | 51 | 11.56 | 0.46 | -0.72 |
| DT18 | 42.7±3.8 | 34 | 43 | 56 | 8.89 | 0.19 | 0.11 |
| NJ18 | 40.8±4.0 | 31 | 41 | 55 | 9.76 | 0.52 | 0.45 |
| YC18 | 48.0±7.7 | 31 | 46 | 67 | 16.15 | 0.73 | -0.03 |
| XX18 | 48.3±8.0 | 37 | 46 | 67 | 16.49 | 1.00 | 0.13 |
| BLUP | 43.5±3.4 | 35 | 43 | 53 | 7.78 | 0.36 | -0.16 |
| 注: 前10行数据来自E-FT数据集; BLUP表示B-FT数据集的播种至开花时间(d)。 Note: The first 10 rows of data belong to E-FT data set; BLUP represents the days from sowing to flowering of B-FT data set. | |||||||
所有环境下, 花期实测值和BLUP值的偏度变化为0.15~1.00, 峰度变化为-0.72~2.57, 表明, 花期实测性状和BLUP值均呈现近似的正态分布, 具有一定的数量性状特征。
2.2 群体结构、亲缘关系与PCA主成分分析利用60 712个SNP标记分析183个供试大豆品种的群体结构。当K=3时, ΔK最大, 表明应将供试大豆群体分为3个亚群(图 1-A); 主成分分析(principal components analysis, PCA)结果也表明供试大豆可分为3类(图 1-B); 依照基因型数据所计算的遗传距离, 按Neighbor-joining进行聚类分析, 所有材料可分为3类(图 1-C)。3个分析结果可进行相互验证, 均可将183个供试大豆材料分为3个亚群, Sub-pop1、2、3分别包含61、64、58个供试大豆品种, 3个亚群花期性状平均值分别为41、44和47 d, 组间差异显著(P < 0.001)(图 1-D、E)。分析结果表明当亚群数为3时, 能将供试大豆进行较好分类, 因此, 本研究选取K=3时的Q矩阵作为后续多种GWAS分析时的协变量, 用于下文的研究。

|
图 1 183个供试大豆品种的群体结构分析结果 Fig. 1 Population structure of 183 soybean cultivars A. 根据Evanno等[20]方法计算的供试材料群体结构K值; B. 供试材料的主成分分析图, 以2个维度来展示群体分层情况; C. 供试材料的NJ-Tree聚类分析结果; D. 183个大豆品种的群体划分, 不同颜色代表不同的亚群; E. 亚群间花期性状差异的显著性分析, ***P < 0.001。 A. Calculation of the true K of the 183 soybean population following procedure outlined by Evanno et al[20]; B. PCA plot of the 183 accessions, two-dimensional scales were used to reveal population stratification; C. A Neighbor-joining tree of the tested accessions; D. Population structure of 183 soybean cultivars based on SNP markers. Three colors represent three subpopulations, respectively; E. Significant analysis of the differences in the flowering time trait among three sub-pops, *** P < 0.001. |
为了评价模型品种参数校准的结果, 将2组品种参数代入CROPGRO大豆花期模拟模型中运行, 得到E-CP和B-CP在10个环境下的大豆花期模拟结果。
通过分析花期数据集与对应模拟花期的1∶1图(图 2-a), 发现大多环境下模拟值与实测值较好分布在对角线的两侧。B-CP和E-CP的大豆花期模拟结果在10个环境下的ARMSE分别为1.65和3.47 d。进一步分析B-CP的结果(图 2-b), 可以看出对LS11和XX18的模拟效果较差, 实测值分别集中在对角线的两端, 这可能是由于XX18相比其他环境开花迟, 且LS11相比其他环境开花更早导致的。进一步分析拟合情况较差的品种, 发现这些品种的花期大多与其余品种存在较大差异。比较不同地点平均RMSE, 可以看出E-CP的RMSE为2.47~5.14 d, B-CP为0.52~3.74 d, B-CP的模型精度明显高于E-CP。因此, 在后续的模型品种参数遗传解析研究中, 仅将花期BLUP值和基于BLUP值分解的模型品种参数作为全基因组关联分析的目标性状。

|
图 2 花期数据与模拟花期的模型模拟结果 Fig. 2 Flowering data and model simulation results of simulated flowering a. E-FT数据集的模型运行结果; b. B-FT数据集的模拟模型运行结果。 a. The running result of the model of the E-FT dataset; b. The running result of the simulation model of the B-FT dataset. |
进一步对B-CP品种参数集进行统计分析。为表述方便, 将由B-FT计算所得的CSDL、PPSEN和EMFL 3个品种参数分别命名为B-CSDL、B-PPSEN和B-EMFL。从表 3可见: 不同品种的B-CSDL、B-PPSEN、B-EMFL的参数值分别为11.79~14.38、0.13~0.30、25.49~28.89, 变异系数分别为4.38%、12.59%、2.62%。B-CSDL、B-PPSEN和B-EMFL的极差分别为2.82、0.26和3.40, 分别占品种参数取值范围的91.84%、64.84%和25.37%, 可见B-CSDL和B-PPSEN广泛分布在事先设置的品种参数取值范围内。从正态性分析来看, B-CSDL和B-EMFL更为正态, B-PPSEN的偏度为5.64, 峰度为42.14, 说明该参数左偏且有尖峰。以上分析表明: B-CSDL相对于B-PPSEN和B-EMFL有更好的正态性。
| 品种参数 Cultivar parameters |
均值±标准差 Mean±SD |
最小值 Minimum |
50%分位数 50% quantile |
最大值 Maximum |
变异系数/% CV |
偏度 Skewness |
峰度 Kurtosis |
| B-CSDL | 12.420±0.540 | 11.790 | 12.270 | 14.380 | 4.38 | 1.17 | 1.08 |
| B-PPSEN | 0.140±0.018 | 0.129 | 0.135 | 0.295 | 12.59 | 5.64 | 42.14 |
| B-EMFL | 28.080±0.740 | 25.490 | 28.280 | 28.890 | 2.62 | -1.19 | 0.94 |
基于Q+K混合线性模型, 利用7种GWAS分析方法, 以大豆花期BLUP值和3个CROPGRO大豆花期模拟模型品种参数(B-FT和B-CP)性状与60 172个SNP进行关联分析(图 3), 以-lg p≥3.5为建议阈值(MLM)和建议的LOD≥3.0作为SNP显著关联的标准(其余6种GWAS方法), 共筛选出10、5、1和6个QTN与CSDL、PPSEN、EMFL和花期BLUP性状显著相关(表 4), 这些位点分别位于2、4、5、6、7、8、9、11、14、16、18、19和20号染色体上, 6和16号染色体上的显著关联QTN数最多, 均各为3个, 其中16号染色体上的Gm16_6264904位点, 6号染色体的Gm06_50296444位点, 5号染色体的Gm05_36831206位点以及11号染色体的Gm11_33036496位点分别对CSDL、PPSEN、EMFL和花期BLUP性状具有最大的变异解释率。

|
图 3 CROPGRO大豆花期模拟模型分解参数的曼哈顿图
Fig. 3
A—D代表不同的关联性状。 红线代表了MLM的建议阈值-lg p=3.5。 A-D represent different traits: A. B-FT; B. B-CSDL; C. B-PPSEN; D. B-EMFL. The red line represents the recommended threshold of GWAS-lg p=3.5. |
| 性状 Traits |
标记 Marker |
方法a Methods |
染色体 Chromosome |
位置/bp Position |
LOD值b LOD score |
解释率/%c Explanation rate |
最小等位基因型频率 MAF |
等位基因 Allele |
已报道的标记/QTL/基因d Known marker/QTL/genes |
| B_CSDL | Gm02_4504945 | 1/5 | 2 | 4 504 945 | 4.264 | 7.741 | 0.138 | G/A | First flower 2-g2 |
| Gm04_3068189 | 1/4/6 | 4 | 3 068 189 | 5.980 | 4.960 | 0.164 | C/G | qST-C1-1[33] | |
| Gm06_17601986 | 2/7 | 6 | 17 601 986 | 4.375 | 10.310 | 0.149 | A/C | CDF3[34] | |
| Gm06_33315421 | 4/6 | 6 | 33 315 421 | 5.691 | 4.937 | 0.115 | C/T | R8 full maturity 8-g2 | |
| Gm09_36933334 | 1/4/5/6 | 9 | 36 933 334 | 6.487 | 9.379 | 0.055 | A/T | Pod maturity 5-1 | |
| Gm14_49318529e | 1/4/6 | 14 | 49 318 529 | 6.794 | 6.523 | 0.077 | C/T | ||
| Gm16_6264904 | 4/5/6 | 16 | 6 264 904 | 4.936 | 19.221 | 0.122 | C/T | First flower 4-g63 | |
| Gm16_33562974 | 1/3/6 | 16 | 33 562 974 | 4.134 | 2.984 | 0.492 | A/T | Photoperiod insensitivity 5-4 | |
| Gm18_3908894 | 1/2/3 | 18 | 3 908 894 | 5.883 | 17.990 | 0.448 | C/T | First flower 4-g69 | |
| Gm20_44570183 | 1/3 | 20 | 44 570 183 | 5.147 | 6.823 | 0.137 | G/T | First flower 6-g7 | |
| B_PPSEN | Gm05_5693713 | 3/4 | 5 | 5 693 713 | 9.887 | 0.824 | 0.055 | T/C | |
| Gm06_50296444 | 4/6/7 | 6 | 50 296 444 | 11.373 | 11.024 | 0.077 | A/G | Reproductive stage length 1-g1.2 | |
| Gm07_1381149 | 4/5 | 7 | 1 381 149 | 8.205 | 2.246 | 0.268 | A/T | ||
| Gm08_39656100 | 3/4 | 8 | 39 656 100 | 11.005 | 0.839 | 0.060 | A/G | ||
| Gm09_7957285 | 3/4 | 9 | 7 957 285 | 6.545 | 0.424 | 0.202 | T/C | ||
| B_EMFL BLUP | Gm05_36831206 | 3/5/6/7 | 5 | 36 831 206 | 5.964 | 9.938 | 0.459 | A/G | |
| Gm02_4874346 | 1/4 | 2 | 4 874 346 | 5.880 | 6.383 | 0.071 | A/C | First flower 2-g2 | |
| Gm11_1218676 | 3/4 | 11 | 1 218 676 | 6.538 | 7.391 | 0.470 | T/C | First flower 4-g39 | |
| Gm11_33036496 | 1/3/6 | 11 | 33 036 496 | 9.127 | 9.397 | 0.060 | G/A | Gm11_33034954[31] | |
| Gm16_33562974 | 1/4 | 16 | 33 562 974 | 4.404 | 4.766 | 0.492 | A/T | Photoperiod insensitivity 5-4 | |
| Gm17_6777393 | 5/6 | 17 | 6 777 393 | 4.966 | 12.531 | 0.105 | A/G | AGL8[34] | |
| Gm19_44450022 | 1/3/6 | 19 | 44 450 022 | 3.359 | 2.753 | 0.082 | C/A | First flower 6-3 | |
| 注: a: 1~7分别对应7种不同的GWAS方法(1. MLM; 2. mrMLM; 3. FASTmrMLM; 4. pLARmEB; 5. pKWmEB; 6. ISIS EM-BLASSO; 7. FASTmrEMMA); b, c: 对于同一QTN标记, 仅展示不同GWAS分析方法的LOD值和表型变异解释率最高的数据; d: 被报道的标记、QTL或基因位于显著位点的QTL区域。 Note: a: 1-7 correspond to 7 different GWAS methods(1. MLM; 2. mrMLM; 3. FASTmrMLM; 4. pLARmEB; 5. pKWmEB; 6. ISIS EM-BLASSO; 7. FASTmrEMMA); b, c: For the same QTN, only the data with the highest LOD score and explanation rate of phenotypic variation under multiple GWAS models are displayed; d: Markers reported, QTL or genes are located in the QTL region of the significant QTN. MDF: Minimum allele frequence. | |||||||||
在CSDL参数性状的关联分析结果中, 显著关联QTN的LOD值为4.264~6.487, 解释了2.984%~19.221%的参数表型变异, 9号染色体上的Gm09_36933334位点被4个GWAS分析方法检测到, 同时拥有最高的LOD值, 可解释9.379%的参数表型变异, 与豆荚成熟期相关的QTL(Pod maturity 5-1)[29]在该标记附近(上下500 kb区间)被筛选到。
与PPSEN参数显著关联的QTN的LOD值为6.545~11.373, 可解释0.424%~11.024%的PPSEN表型变异, 其中6号染色体的Gm06_50296444位点可同时被3个GWAS分析方法检测到, 同时拥有最高的LOD值和表型变异解释率, 并在该标记附近筛选出与生殖生长时期长度相关的QTL(Reproductive stage length 1-g1.2)[30]。仅有1个QTN与EMFL参数性状显著相关, 该标记可同时被4种GWAS分析方法检测到, LOD值为5.964。
在花期BLUP值的关联分析结果中, 6个显著关联的QTN的LOD值为3.359~9.124, 可解释2.753%~12.531%的花期表型变异。11号染色体上的Gm11_33036496位点能同时被3个GWAS分析方法检测到, 与该标记位置接近的Gm11_33034954(相距1.542 kb)位点为前人研究中与不同光温条件下大豆花期性状显著关联的SNP位点[31]。此外, 与始花期性状相关的QTL(First flower 6-3)[32]在19号染色体上的Gm19_44450022位点的QTL区间被筛选到, 该位点可同时被3个GWAS分析方法检测到, LOD值为3.359, 并有2.753%的表型变异解释率。
在所有22个显著相关QTN附近, 筛选出16个(72.73%)在前人研究报道中与大豆开花时间、光周期敏感性、生殖生长时期、豆荚成熟期等性状显著相关的SNP标记、QTL或者基因, 说明CROPGRO大豆花期模型品种参数具有一定的遗传特征。同时将其余6个QTN(Gm14_49318529、Gm05_5693713、Gm07_1381149、Gm08_39656100、Gm09_7957285和Gm05_36831206)作为候选位点, 对其上下500 kb区间的候选基因功能进行预测。
2.5 候选基因功能预测根据显著关联QTN和连锁不平衡区间确定与大豆开花、光周期响应等相关的候选位点, 并对位点内的候选基因功能进行预测。其中, Glyma05g06220(与Gm05_5693713相距264.014 kb)与LEUNIG_HOMOLOG(LUH)基因同源, 该基因为拟南芥花同源异型基因AGAMOUS(AG)的转录抑制因子, LUH基因会与拟南芥SEUSS(SEU)基因相互作用, 共同调控拟南芥花的发育[35-36]; Glyma05g31710(与Gm05_36831206相距24.422 kb)与拟南芥花器官建成基因SEPALLATA(SEP)[37]同源, 该基因的表达水平与外界的光照强度呈显著正相关[38]; 在距Gm07_1381149位点347.120 kb的位置上筛选出的Glyma07g01601是拟南芥早花基因EARLY FLOWERING 3(ELF3)的同源基因, 该基因是拟南芥生物钟调控的重要组件, 对光反应敏感, 具有在夜间抑制拟南芥生长的功能[39-40]; 位于Gm08_39656100位点374.554 kb位置的Glyma08g40330基因是拟南芥细胞分裂素信号通路反应调节因子ARABIDOPSIS TYPE A RESPONSE REGULATOR-3(ARR3)的同源基因, ARR3基因除了响应细胞分裂素以外, 还参与调节生物钟, 对拟南芥昼夜节律具有调控作用[41]; 另一个基因Glyma14g40030位于显著关联QTN Gm14_49318529的253.231 kb的距离上, 该基因与拟南芥S期激酶相关蛋白基因S-PHASE KINASEASSOCIA TED PROTEIN1(SKP1)同源, SKP1蛋白通常与CDC53P/Cull F-box形成SCF复合体参与泛素蛋白降解途径调控细胞循环、生物节律以及雄性不育等许多重要反应[42], 该基因在茎尖、花序和花原基等分裂旺盛的组织中高水平表达[43]。
3 讨论 3.1 BLUP数据集选择依据BLUP方法是由Henderson[44]针对不平衡资料提出的育种值预测方法, 通过该方法可以有效消除各种非遗传因素的影响, 实现对育种值的最佳线性无偏估计, 从而提高选择的准确性。目前已在大豆、高粱、水稻等多种作物的农艺性状GWAS分析中得到广泛应用。
CROPGRO大豆花期模拟模型引入了品种参数来反映品种间的遗传差异, 但是事实上, 大多数作物生长模型建立之初并没有考虑品种参数的潜在遗传基础[8]。前人研究[8]证明: 将已校正好的作物模型应用到新环境时, 往往需要重新校正模型品种参数。这说明品种参数并非如同建模者所设想的那样, 完全由品种自身的基因型特异性决定, 更有可能是一个综合基因与环境互作的参数。本研究希望获取能够反映品种自身遗传学特性的模型品种参数, 尽可能减少品种参数的环境效应, 因此选用BLUP花期值过滤掉实测花期的环境效应。综上所述, 利用排除一定环境效应的BLUP值进行模型品种参数分解是更为合理的参数校正方式。
3.2 CROPGRO大豆花期模拟模型品种参数的基因定位分析本研究基于183个江淮大豆品种, 利用60 712个SNP标记对大豆BLUP花期和3个品种参数进行全基因组关联分析。共采用了7种全基因组关联分析方法, 不同GWAS方法分析原理不同, 各种方法之间互相弥补, 有效降低了只有1种分析方法造成的定位结果假阳性率。在检测结果中, 以-lg p≥3.5和的LOD≥3.0作为建议阈值, 筛选出在7种方法间至少被2次重复检测到的QTN位点。最终得到与CSDL、PPSEN、EMFL和花期BLUP性状相关的可信QTN分别为10、5、1和6个。
在本研究中, 模型品种参数定位到的显著位点与BLUP花期定位到的位点位置差异很大, 两者仅有1个共同定位的显著位点(Gm16_33562974)。类似的结果同样出现在Gu等[45]、Guitton等[10]、Dingkuhn等[11]的研究中, 他们均发现模型品种参数能够定位到更多且显著性更高的位点, 并且与表型定位结果的重复度很低, 但都未对这一现象作出解释。针对本研究的结果, 我们认为模型品种参数作为实测性状分解的组分, 在数量上往往多于原始的实测性状, 所以可以定位到更多位点; 品种参数反映了不同环境效应对大豆品种花期的影响, 这可能是实测性状和品种参数定位结果差异大、共同定位少的原因。
本研究参考Dingkuhn等[11]的研究, 使用生长模型模拟花期的精度以及品种参数的定位结果来作为CROPGROW大豆花期模拟模型的品种参数是否具有遗传可解释性的两个评价指标。前者用于评价模型对当地生态环境的适用性, 后者则要求品种参数定位的显著位点能与已知的位点或基因位置重复, 只有这样才能表明本研究的品种参数具有生物学意义。在B-CSDL的定位结果中, 共找到10个候选基因, 其中9个已在大豆中被证明与花期相关, 其余1个候选基因在比对拟南芥数据库中发现与SKP1同源, 该基因在茎尖、花序和花原基等分裂旺盛的组织中高水平表达。在B-PPSEN参数的定位结果中, 共筛选出5个候选基因, 其中1个已验证与大豆花期相关, 其余4个候选基因中的3个与拟南芥光反应、昼夜节律或花的发育有关, 分别为ELF3、ARR3和LUH基因。在B-EMFL参数的定位结果中, 只找到1个候选基因, 该基因在比对拟南芥数据库后发现与SEP基因同源, 该基因的表达水平与外界的光照强度呈现显著的正相关。
综上所述, CROPGRO大豆花期模拟模型品种参数CSDL、PPSEN具有遗传学上的解释, 品种参数EMFL在遗传学上的解释较弱。所以, 即使物候学模型简化了生物学过程, 并将先验理论强加于数据之上, 但是模型品种参数还是能体现出它的遗传特性。利用模型品种参数定位到的显著SNP标记, 可提高原CROPGROW大豆花期模拟模型品种参数的遗传可解释性, 促进生理学与基因组学联系起来。
| [1] |
Holzworth D P, Huth N I, de Voil P G, et al. APSIM-Evolution towards a new generation of agricultural systems simulation[J]. Environmental Modelling & Software, 2014, 62: 327-350. |
| [2] |
Wang E L, Brown H E, Rebetzke G J, et al. Improving process-based crop models to better capture genotype×environment×management interactions[J]. Journal of Experimental Botany, 2019, 70(9): 2389-2401. DOI:10.1093/jxb/erz092 |
| [3] |
Robertson M J, Rebetzke G J, Norton R M. Assessing the place and role of crop simulation modelling in Australia[J]. Crop and Pasture Science, 2015, 66(9): 877-893. DOI:10.1071/CP14361 |
| [4] |
Yin X Y, Chasalow S D, Dourleijn J C, et al. Coupling estimated effects of QTLs for physiological traits to a crop growth model: predicting yield variation among recombinant inbred lines in barley[J]. Heredity, 2000, 85(6): 539-549. DOI:10.1046/j.1365-2540.2000.00790.x |
| [5] |
White J W, Hoogenboom G. Simulating effects of genes for physiological traits in a process-oriented crop model[J]. Agronomy Journal, 1996, 88(3): 416-422. DOI:10.2134/agronj1996.00021962008800030009x |
| [6] |
Boote K J, Vallejos C E, Jones J W, et al. Crop modeling approaches for predicting phenotype of grain legumes with linkage to genetic information[M]//Crop Systems Biology. Switzerland: Springer International Publishing, 2016: 163-192.
|
| [7] |
Tardieu F. Virtual plants: modelling as a tool for the genomics of tolerance to water deficit[J]. Trends in Plant Science, 2003, 8(1): 9-14. DOI:10.1016/S1360-1385(02)00008-0 |
| [8] |
Yin X Y, van der Linden C G, Struik P C. Bringing genetics and biochemistry to crop modelling, and vice versa[J]. European Journal of Agronomy, 2018, 100: 132-140. DOI:10.1016/j.eja.2018.02.005 |
| [9] |
Mavromatis T, Boote K J, Jones J W, et al. Developing genetic coefficients for crop simulation models with data from crop performance trials[J]. Crop Science, 2001, 41(1): 40-51. DOI:10.2135/cropsci2001.41140x |
| [10] |
Guitton B, Théra K, Tékété M L, et al. Integrating genetic analysis and crop modeling: a major QTL can finely adjust photoperiod-sensitive Sorghum flowering[J]. Field Crops Research, 2018, 221: 7-18. DOI:10.1016/j.fcr.2018.02.007 |
| [11] |
Dingkuhn M, Pasco R, Pasuquin J M, et al. Crop-model assisted phenomics and genome-wide association study for climate adaptation of indica rice. 1. phenology[J]. Journal of Experimental Botany, 2017, 68(15): 4369-4388. DOI:10.1093/jxb/erx249 |
| [12] |
Kadam N N, Jagadish S V K, Struik P C, et al. Incorporating genome-wide association into eco-physiological simulation to identify markers for improving rice yields[J]. Journal of Experimental Botany, 2019, 70(9): 2575-2586. DOI:10.1093/jxb/erz120 |
| [13] |
Fehr W R, Caviness C E, Burmood D T, et al. Stage of development descriptions for soybeans, Glycine max(L.) Merrill[J]. Crop Science, 1971, 11(6): 929-931. DOI:10.2135/cropsci1971.0011183X001100060051x |
| [14] |
Bates D, Mächler M, Bolker B M, et al. Fitting linear mixed-effects models using lme4[J]. Journal of Statistical Software, 2015, 67(1): 133-199. |
| [15] |
Li L H, Guo N, Niu J P, et al. Loci and candidate gene identification for resistance to Phytophthora sojae via association analysis in soybean[Glycine max(L.) Merr.][J]. Molecular Genetics and Genomics, 2016, 291(3): 1095-1103. DOI:10.1007/s00438-015-1164-x |
| [16] |
Jones J W, Hoogenboom G, Porter C H, et al. The DSSAT cropping system model[J]. European Journal of Agronomy, 2003, 18(3/4): 235-265. |
| [17] |
White J W, Hoogenboom G, Jones J W, et al. Evaluation of the dry bean model Beangro V1.01 for crop production research in a tropical environment[J]. Experimental Agriculture, 1995, 31(2): 241-254. DOI:10.1017/S001447970002531X |
| [18] |
Tsuji G Y, Hoogenboom G, Thornton P K. Understanding Options for Agricultural Production[M]. Dordrecht, Netherlands: Springer, 1998.
|
| [19] |
He J Q, Jones J W, Graham W D, et al. Influence of likelihood function choice for estimating crop model parameters using the generalized likelihood uncertainty estimation method[J]. Agricultural Systems, 2010, 103(5): 256-264. DOI:10.1016/j.agsy.2010.01.006 |
| [20] |
Evanno G, Regnaut S, Goudet J. Detecting the number of clusters of individuals using the software STRUCTURE: a simulation study[J]. Molecular Ecology, 2005, 14(8): 2611-2620. DOI:10.1111/j.1365-294X.2005.02553.x |
| [21] |
Bradbury P J, Zhang Z W, Kroon D E, et al. TASSEL: software for association mapping of complex traits in diverse samples[J]. Bioinformatics, 2007, 23(19): 2633-2635. DOI:10.1093/bioinformatics/btm308 |
| [22] |
Wang S B, Feng J Y, Ren W L, et al. Improving power and accuracy of genome-wide association studies via a multi-locus mixed linear model methodology[J]. Scientific Reports, 2016, 6: 19444. DOI:10.1038/srep19444 |
| [23] |
Tamba C L, Ni Y L, Zhang Y M. Iterative sure independence screening EM-Bayesian LASSO algorithm for multi-locus genome-wide association studies[J]. PLoS Computational Biology, 2017, 13(1): e1005357. DOI:10.1371/journal.pcbi.1005357 |
| [24] |
Wen Y J, Zhang H W, Ni Y L, et al. Methodological implementation of mixed linear models in multi-locus genome-wide association studies[J]. Briefings in Bioinformatics, 2018, 19(4): 700-712. DOI:10.1093/bib/bbw145 |
| [25] |
Zhang J, Feng J Y, Ni Y L, et al. pLARmEB: integration of least angle regression with empirical Bayes for multilocus genome-wide association studies[J]. Heredity, 2017, 118(6): 517-524. DOI:10.1038/hdy.2017.8 |
| [26] |
Ren W L, Wen Y J, Dunwell J M, et al. pKWmEB: integration of Kruskal-Wallis test with empirical Bayes under polygenic background control for multi-locus genome-wide association study[J]. Heredity, 2018, 120(3): 208-218. DOI:10.1038/s41437-017-0007-4 |
| [27] |
Zhang Z W, Ersoz E, Lai C Q, et al. Mixed linear model approach adapted for genome-wide association studies[J]. Nature Genetics, 2010, 42(4): 355-360. DOI:10.1038/ng.546 |
| [28] |
Brodie A, Azaria J R, Ofran Y. How far from the SNP may the causative genes be?[J]. Nucleic Acids Research, 2016, 44(13): 6046-6054. DOI:10.1093/nar/gkw500 |
| [29] |
Lee S H, Bailey M A, Mian M A R, et al. Identification of quantitative trait loci for plant height, lodging, and maturity in a soybean population segregating for growth habit[J]. Theoretical and Applied Genetics, 1996, 92(5): 516-523. DOI:10.1007/BF00224553 |
| [30] |
Fang C, Ma Y, Wu S, et al. Genome-wide association studies dissect the genetic networks underlying agronomical traits in soybean[J]. Genome Biology, 2017, 18(1): 161-173. DOI:10.1186/s13059-017-1289-9 |
| [31] |
Mao T T, Li J Y, Wen Z X, et al. Association mapping of loci controlling genetic and environmental interaction of soybean flowering time under various photo-thermal conditions[J]. BMC Genomics, 2017, 18(1): 415-431. |
| [32] |
Orf J H, Chase K, Jarvik T, et al. Genetics of soybean agronomic traits: I. comparison of three related recombinant inbred populations[J]. Crop Science, 1999, 39(6): 1642-1651.
|
| [33] |
宁海龙, 庄煦, 杨畅, 等. 大豆四向重组自交系群体生育进程稳定QTL定位[J]. 大豆科学, 2017, 36(5): 692-698. Ning H L, Zhuang X, Yang C, et al. Mapping stable QTLs for growth period procedure based on four-way recombination inbred line population[J]. Soybean Science, 2017, 36(5): 692-698 (in Chinese with English abstract). |
| [34] |
Kim M Y, Shin J H, Kang Y J, et al. Divergence of flowering genes in soybean[J]. Journal of Biosciences, 2012, 37(5): 857-870. DOI:10.1007/s12038-012-9252-0 |
| [35] |
Alvarez-Buylla E R, Benítez M, Corvera-Poiré A, et al. Flower development[M]//The Arabidopsis Book. The American Society of Plant Biologists, 2010: e0127.
|
| [36] |
Sitaraman J, Bui M, Liu Z C. LEUNIG_HOMOLOG and LEUNIG perform partially redundant functions during Arabidopsis embryo and floral development[J]. Plant Physiology, 2008, 147(2): 672-681. DOI:10.1104/pp.108.115923 |
| [37] |
Krizek B A, Fletcher J C. Molecular mechanisms of flower development: an armchair guide[J]. Nature Reviews Genetics, 2005, 6(9): 688-698. |
| [38] |
Heddad M, Adamska I. Light stress-regulated two-helix proteins in Arabidopsis thaliana related to the chlorophyll a/b-binding gene family[J]. Proc Natl Acad USA, 2000, 97(7): 3741-3746. |
| [39] |
Anwer M U, Davis A, Davis S J, et al. Photoperiod sensing of the circadian clock is controlled by EARLY FLOWERING 3 and GIGANTEA[J]. The Plant Journal, 2020, 101(6): 1397-1410. DOI:10.1111/tpj.14604 |
| [40] |
Kanesaka Y, Okada M, Ito S, et al. Monitoring single-cell bioluminescence of Arabidopsis leaves to quantitatively evaluate the efficiency of a transiently introduced CRISPR/Cas9 system targeting the circadian clock gene ELF3[J]. Plant Biotechnology, 2019, 36(3): 187-193. |
| [41] |
Ishida K, Yamashino T, Mizuno T. Expression of the cytokinin-induced type-A response regulator gene ARR9 is regulated by the circadian clock in Arabidopsis thaliana[J]. Bioscience, Biotechnology, and Biochemistry, 2008, 72(11): 3025-3029. |
| [42] |
Craig K L, Tyers M. The F-box: a new motif for ubiquitin dependent proteolysis in cell cycle regulation and signal transduction[J]. Progress in Biophysics and Molecular Biology, 1999, 72(3): 299-328. |
| [43] |
Porat R, Lu P Z, O'Neill S D. Arabidopsis SKP1, a homologue of a cell cycle regulator gene, is predominantly expressed in meristematic cells[J]. Planta, 1998, 204(3): 345-351. |
| [44] |
Henderson C R. Selection index and expected genetic advance[J]. Statistical Genetics and Plant Breeding, 1963, 982: 141-163. |
| [45] |
Gu J F, Yin X Y, Zhang C W, et al. Linking ecophysiological modelling with quantitative genetics to support marker-assisted crop design for improved yields of rice(Oryza sativa) under drought stress[J]. Annals of Botany, 2014, 114(3): 499-511. |